The RDKit Book¶
Misc Cheminformatics Topics¶
Aromaticity¶
Aromaticity is one of those unpleasant topics that is simultaneously simple and impossibly complicated. Since neither experimental nor theoretical chemists can agree with each other about a definition, it’s necessary to pick something arbitrary and stick to it. This is the approach taken in the RDKit.
Instead of using patterns to match known aromatic systems, the aromaticity perception code in the RDKit uses a set of rules. The rules are relatively straightforward.
Aromaticity is a property of atoms and bonds in rings. An aromatic bond must be between aromatic atoms, but a bond between aromatic atoms does not need to be aromatic.
For example the fusing bonds here are not considered to be aromatic by the RDKit:
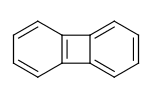
>>> from rdkit import Chem
>>> m = Chem.MolFromSmiles('C1=CC2=C(C=C1)C1=CC=CC=C21')
>>> m.GetAtomWithIdx(3).GetIsAromatic()
True
>>> m.GetAtomWithIdx(6).GetIsAromatic()
True
>>> m.GetBondBetweenAtoms(3,6).GetIsAromatic()
False
The RDKit supports a number of different aromaticity models and allows the user to define their own by providing a function that assigns aromaticity.
The RDKit Aromaticity Model¶
A ring, or fused ring system, is considered to be aromatic if it obeys the 4N+2 rule. Contributions to the electron count are determined by atom type and environment. Some examples:
Fragment |
Number of pi electrons |
c(a)a |
1 |
n(a)a |
1 |
An(a)a |
2 |
o(a)a |
2 |
s(a)a |
2 |
se(a)a |
2 |
te(a)a |
2 |
O=c(a)a |
0 |
N=c(a)a |
0 |
*(a)a |
0, 1, or 2 |
Notation a: any aromatic atom; A: any atom, include H; *: a dummy atom
Notice that exocyclic bonds to electronegative atoms “steal” the valence electron from the ring atom and that dummy atoms contribute whatever count is necessary to make the ring aromatic.
The use of fused rings for aromaticity can lead to situations where individual rings are not aromatic, but the fused system is. An example of this is azulene:
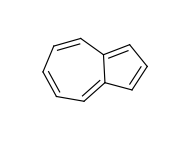
An extreme example, demonstrating both fused rings and the influence of exocyclic double bonds:
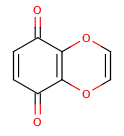
>>> m=Chem.MolFromSmiles('O=C1C=CC(=O)C2=C1OC=CO2')
>>> m.GetAtomWithIdx(6).GetIsAromatic()
True
>>> m.GetAtomWithIdx(7).GetIsAromatic()
True
>>> m.GetBondBetweenAtoms(6,7).GetIsAromatic()
False
A special case, heteroatoms with radicals are not considered candidates for aromaticity:
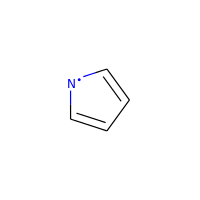
>>> m = Chem.MolFromSmiles('C1=C[N]C=C1')
>>> m.GetAtomWithIdx(0).GetIsAromatic()
False
>>> m.GetAtomWithIdx(2).GetIsAromatic()
False
>>> m.GetAtomWithIdx(2).GetNumRadicalElectrons()
1
Charged carbons with radicals are also not considered:
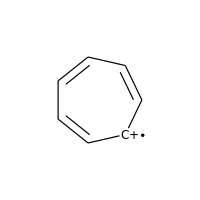
>>> m = Chem.MolFromSmiles('C1=CC=CC=C[C+]1')
>>> m.GetAtomWithIdx(0).GetIsAromatic()
False
>>> m.GetAtomWithIdx(6).GetIsAromatic()
False
>>> m.GetAtomWithIdx(6).GetFormalCharge()
1
>>> m.GetAtomWithIdx(6).GetNumRadicalElectrons()
1
Neutral carbons with radicals, however, are still considered:
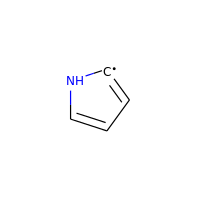
>>> m = Chem.MolFromSmiles('C1=[C]NC=C1')
>>> m.GetAtomWithIdx(0).GetIsAromatic()
True
>>> m.GetAtomWithIdx(1).GetIsAromatic()
True
>>> m.GetAtomWithIdx(1).GetNumRadicalElectrons()
1
The Simple Aromaticity Model¶
This one is quite simple: only five- and six-membered simple rings are considered candidates for aromaticity. The same electron-contribution counts listed above are used.
The MDL Aromaticity Model¶
This isn’t well documented (at least not publicly), so we tried to reproduce what’s provided in the oechem documentation (https://docs.eyesopen.com/toolkits/python/oechemtk/aromaticity.html)
fused rings (i.e. azulene) can be aromatic
five-membered rings are not aromatic (though they can be part of fused aromatic systems)
only C and N can be aromatic
only one electron donors are accepted
atoms with exocyclic double bonds are not aromatic
Note: For reasons of computational expediency, aromaticity perception is only done for fused-ring systems where all members are at most 24 atoms in size.
SMILES Support and Extensions¶
The RDKit covers all of the standard features of Daylight SMILES [2] as well as some useful extensions.
Here’s the (likely partial) list of extensions:
Aromaticity¶
te (aromatic Te) is accepted. Here is an example with tellurophene-2-carboxylic acid:
>>> m = Chem.MolFromSmiles('OC(=O)c1[te]ccc1')
>>> m.GetAtomWithIdx(4).GetIsAromatic()
True
Dative bonds¶
<- and -> create a dative bond between the atoms, direction does matter.
Here’s an example of a bipy-copper complex:
>>> bipycu = Chem.MolFromSmiles('c1cccn->2c1-c1n->3cccc1.[Cu]23(Cl)Cl')
>>> bipycu.GetBondBetweenAtoms(4,12).GetBondType()
rdkit.Chem.rdchem.BondType.DATIVE
>>> Chem.MolToSmiles(bipycu)
'[Cl][Cu]1([Cl])<-[n]2ccccc2-c2cccc[n]->12'
Dative bonds have the special characteristic that they don’t affect the valence on the start atom, but do affect the end atom. So in this case, the N atoms involved in the dative bond have the valence of 3 that we expect from bipy, while the Cu has a valence of 4:
>>> bipycu.GetAtomWithIdx(4).GetTotalValence()
3
>>> bipycu.GetAtomWithIdx(12).GetTotalValence()
4
Ring closures¶
%(N) notation is supported for ring closures, where N is a single digit %(N) up to
five digits %(NNNNN). Here is an example:
>>> m = Chem.MolFromSmiles('C%(1000)OC%(1000)')
>>> m.GetAtomWithIdx(0).IsInRing()
True
>>> m.GetAtomWithIdx(2).IsInRing()
True
Specifying atoms by atomic number¶
The [#6] construct from SMARTS is supported in SMILES.
Quadruple bonds¶
The token $ can be used to represent quadruple bonds in SMILES and SMARTS.
CXSMILES/CXSMARTS extensions¶
The RDKit supports parsing and writing a subset of the extended SMILES/SMARTS functionality introduced by ChemAxon [4].
The features which are parsed include:
atomic coordinates
()atomic values
$_AV:atomic labels/aliases
$(recognized aliases are_AP,star_e,Q_e,QH_p,AH_P,X_p,XH_p,M_p,MH_p,*)atomic properties
atompropcoordinate/dative bonds
C(these are translated into dative bonds)hydrogen bonds
Hzero order bonds bonds
Z(custom extension, same syntax as c/t/ctu below)radicals
^enhanced stereo (these are converted into
StereoGroups)linknodes
LNvariable/multi-center attachments
mring bond count specifications
rbnon-hydrogen substitution count specifications
sunsaturation specification
uwedged bonds (only when atomic coordinates are present):
wU,wDwiggly bonds
wdouble bond stereo (only for ring bonds)
c,t,ctuSGroup Data
SgDpolymer SGroups
SgSGroup Hierarchy
SgH
The features which are written by rdkit.Chem.rdmolfiles.MolToCXSmiles() and
rdkit.Chem.rdmolfiles.MolToCXSmarts()
(note the specialized writer functions) include:
atomic coordinates
atomic values
atomic labels
atomic properties
dative bonds (only if dative bonds are not also being written to the SMILES/SMARTS)
radicals
enhanced stereo
linknodes
wedged bonds (only when atomic coordinates are also written)
wiggly bonds
double bond stereo (only for ring bonds)
SGroup Data
polymer SGroups
SGroup Hierarchy
>>> m = Chem.MolFromSmiles('OC')
>>> m.GetAtomWithIdx(0).SetProp('p1','2')
>>> m.GetAtomWithIdx(1).SetProp('p1','5')
>>> m.GetAtomWithIdx(1).SetProp('p2','A1')
>>> m.GetAtomWithIdx(0).SetProp('atomLabel','O1')
>>> m.GetAtomWithIdx(1).SetProp('atomLabel','C2')
>>> m.GetBondWithIdx(0).SetBondType(Chem.BondType.ZERO)
>>> Chem.MolToCXSmiles(m)
'C~O |$C2;O1$,atomProp:0.p1.5:0.p2.A1:1.p1.2,Z:0|'
Reading molecule names¶
If the SMILES/SMARTS and the optional CXSMILES extensions are followed by whitespace and another string, the SMILES/SMARTS parsers will interpret this as the molecule name:
>>> m = Chem.MolFromSmiles('CO carbon monoxide')
>>> m.GetProp('_Name')
'carbon monoxide'
>>> m2 = Chem.MolFromSmiles('CO |$C2;O1$| carbon monoxide')
>>> m2.GetAtomWithIdx(0).GetProp('atomLabel')
'C2'
>>> m2.GetProp('_Name')
'carbon monoxide'
This can be disabled while still parsing the CXSMILES:
>>> ps = Chem.SmilesParserParams()
>>> ps.parseName = False
>>> m3 = Chem.MolFromSmiles('CO |$C2;O1$| carbon monoxide',ps)
>>> m3.HasProp('_Name')
0
>>> m3.GetAtomWithIdx(0).GetProp('atomLabel')
'C2'
Note that if you disable CXSMILES parsing but pass in a string which includes CXSMILES it will be interpreted as (part of) the name:
>>> ps = Chem.SmilesParserParams()
>>> ps.allowCXSMILES = False
>>> m4 = Chem.MolFromSmiles('CO |$C2;O1$| carbon monoxide',ps)
>>> m4.GetProp('_Name')
'|$C2;O1$| carbon monoxide'
Finally, if you disable parsing of both CXSMILES and names, then extra text in the SMILES/SMARTS string will result in errors: .. doctest:
>>> ps = Chem.SmilesParserParams()
>>> ps.allowCXSMILES = False
>>> ps.parseName = False
>>> m5 = Chem.MolFromSmiles('CO |$C2;O1$| carbon monoxide',ps)
>>> m5 is None
True
>>> m5 = Chem.MolFromSmiles('CO carbon monoxide',ps)
>>> m5 is None
True
The examples in this sectin all used the SMILES parser, but the SMARTS parser behaves the same way.
SMARTS Support and Extensions¶
The RDKit covers most of the standard features of Daylight SMARTS [3] as well as some useful extensions.
Here’s the (hopefully complete) list of SMARTS features that are not supported:
Non-tetrahedral chiral classes
the
@?operatorexplicit atomic masses (though isotope queries are supported)
component level grouping requiring matches in different components, i.e.
(C).(C)
Here’s the (likely partial) list of extensions:
Hybridization queries¶
^0matches S hybridized atoms
^1matches SP hybridized atoms
^2matches SP2 hybridized atoms
^3matches SP3 hybridized atoms
^4matches SP3D hybridized atoms
^5matches SP3D2 hybridized atoms
>> Chem.MolFromSmiles('CC=CF').GetSubstructMatches(Chem.MolFromSmarts('[^2]'))
((1,), (2,))
Dative bonds¶
<- and -> match the corresponding dative bonds, direction does matter.
>>> Chem.MolFromSmiles('C1=CC=CC=N1->[Fe]').GetSubstructMatches(Chem.MolFromSmarts('[#7]->*'))
((5, 6),)
>>> Chem.MolFromSmiles('C1=CC=CC=N1->[Fe]').GetSubstructMatches(Chem.MolFromSmarts('*<-[#7]'))
((6, 5),)
Heteroatom neighbor queries¶
the atom query
zmatches atoms that have the specified number of heteroatom (i.e. not C or H) neighbors. For example,z2would match the second C inCC(=O)O.the atom query
Zmatches atoms that have the specified number of aliphatic heteroatom (i.e. not C or H) neighbors.
>>> Chem.MolFromSmiles('O=C(O)c1nc(O)ccn1').GetSubstructMatches(Chem.MolFromSmarts('[z2]'))
((1,), (3,), (5,))
>>> Chem.MolFromSmiles('O=C(O)c1nc(O)ccn1').GetSubstructMatches(Chem.MolFromSmarts('[Z2]'))
((1,),)
>>> Chem.MolFromSmiles('O=C(O)c1nc(O)ccn1').GetSubstructMatches(Chem.MolFromSmarts('[Z1]'))
((5,),)
Range queries¶
Ranges of values can be provided for many query types that expect numeric values.
The query types that currently support range queries are:
D, h, r, R, v, x, X, z, Z, +, -
- Here are some examples:
D{2-4}matches atoms that have between 2 and 4 (inclusive) explicit connections.D{-3}matches atoms that have less than or equal to 3 explicit connections.D{2-}matches atoms that have at least 2 explicit connections.
>>> Chem.MolFromSmiles('CC(=O)OC').GetSubstructMatches(Chem.MolFromSmarts('[z{1-}]'))
((1,), (4,))
>>> Chem.MolFromSmiles('CC(=O)OC').GetSubstructMatches(Chem.MolFromSmarts('[D{2-3}]'))
((1,), (3,))
>>> Chem.MolFromSmiles('CC(=O)OC.C').GetSubstructMatches(Chem.MolFromSmarts('[D{-2}]'))
((0,), (2,), (3,), (4,), (5,))
SMARTS Reference¶
Note that the text versions of the tables below include some backslash characters to escape special characters. This is a wart from the documentation system we are using. Please ignore those characters.
Atoms¶
Primitive |
Property |
“Default value” |
Range? |
Notes |
|---|---|---|---|---|
a |
“aromatic atom” |
|||
A |
“aliphatic atom” |
|||
d |
“non-hydrogen degree” |
1 |
Y |
extension |
D |
“explicit degree” |
1 |
Y |
|
h |
“number of implicit hs” |
>0 |
Y |
|
H |
“total number of Hs” |
1 |
||
r |
“size of smallest SSSR ring” |
>0 |
Y |
|
R |
“number of SSSR rings” |
>0 |
Y |
|
k |
“size of SSSR ring” |
>0 |
Y |
extension |
v |
“total valence” |
1 |
Y |
|
x |
“number of ring bonds” |
>0 |
Y |
|
X |
“total degree” |
1 |
Y |
|
z |
“number of heteroatom neighbors” |
>0 |
Y |
extension |
Z |
“number of aliphatic heteroatom neighbors” |
>0 |
Y |
extension |
* |
“any atom” |
|||
+ |
“positive charge” |
1 |
Y |
|
++ |
“+2 charge” |
|||
- |
“negative charge” |
1 |
Y |
|
-- |
“-2 charge” |
|||
^0 |
“S hybridized” |
n/a |
N |
extension |
^1 |
“SP hybridized” |
n/a |
N |
extension |
^2 |
“SP2 hybridized” |
n/a |
N |
extension |
^3 |
“SP3 hybridized” |
n/a |
N |
extension |
^4 |
“SP3D hybridized” |
n/a |
N |
extension |
^5 |
“SP3D2 hybridized” |
n/a |
N |
extension |
Bonds¶
Primitive |
Property |
Notes |
|---|---|---|
“” |
“single or aromatic” |
“unspecified bonds” |
- |
single |
|
= |
double |
|
# |
triple |
|
: |
aromatic |
|
~ |
“any bond” |
|
@ |
“ring bond” |
|
/ |
“directional” |
|
\ |
“directional” |
|
-> |
“dative right” |
extension |
<- |
“dative left” |
extension |
Hs in SMARTS¶
Hs in SMARTS are interpreted as hydrogen atoms if the equivalent atom expression would also be a valid SMILES; otherwise they are interpreted as a query for any atom with a single attached hydrogen.
Some examples:
SMARTS |
Interpretation |
|---|---|
[H] |
[#1] |
[H+] |
[#1+] |
[H,Cl] |
[*H1,Cl] |
[HH] |
[*H1;*H1] |
This is somewhat confusing, but is consistent with the Daylight documentation (https://www.daylight.com/dayhtml/doc/theory/theory.smirks.html):
Hence, a single change to SMARTS interpretation, for expressions of the form: [<weight>H<charge><map>]. In SMARTS, these expressions now are interpreted as a hydrogen atom, rather than as any atom with one hydrogen attached. All other SMARTS hydrogen expressions retain their pre-4.51 meanings.
It’s always possible to see the RDKit’s interpretation of a SMARTS using the DescribeQuery() function:
>>> print(Chem.AtomFromSmarts('[H,Cl]').DescribeQuery())
AtomOr
AtomHCount 1 = val
AtomType 17 = val
>>> print(Chem.AtomFromSmarts('[2H+]').DescribeQuery())
AtomAnd
AtomAnd
AtomAtomicNum 1 = val
AtomIsotope 2 = val
AtomFormalCharge 1 = val
The safest (and clearest) way to incorporate H atoms into your queries is to use the atomic number primitive [#1] instead of [H].
Mol/SDF Support and Extensions¶
The RDKit covers an extensive subset of the features in the V2000 and V3000 CTAB specfication. This subset should be better documented.
- Here are the non-element atom queries that are supported:
A: any heavy atom
Q: any non-carbon heavy atom
*: unspecfied (interpreted as any atom)
L: (V2000): atom list
AH: (ChemAxon Extension) any atom
QH: (ChemAxon Extension) any non-carbon atom
X: (ChemAxon Extension) halogen
XH: (ChemAxon Extension) halogen or hydrogen
- M: (ChemAxon Extension) metal (“contains alkali metals, alkaline earth metals, transition
metals, actinides, lanthanides, poor(basic) metals, Ge, Sb, and Po”)
MH: (ChemAxon Extension) metal or hydrogen
- Here’s a partial list of the features that are supported:
enhanced stereochemistry (V3000 only)
Sgroups: Sgroups are read and written, but interpretation of their contents is still very much a work in progress
Dative bonds in V2000 (type 9), despite them not being part of the standard, we support them because they frequently show up in real-world data
Interpretation of the 2D/3D Flag and Stereochemistry¶
Mol blocks can describe 2D or 3D molecules and include a flag (called “dimensional code” in the spec) to indicate whether the coordinates are 2D or 3D. Of course real-world files include every possible combination of 2D/3D flag and 2D/3D coordinates, so we need to decide how to interpret these combinations. Things are made more complicated by the possible presence of wedged bonds in the mol block.
The following table describes how the RDKit interprets all possible combinations of these three variables:
flag |
coords |
wedging |
result |
notes |
|---|---|---|---|---|
2D |
2D |
no |
2D |
no chirality |
3D |
2D |
no |
3D |
no chirality |
3D |
3D |
no |
3D |
chirality from coords |
2D |
3D |
no |
3D |
chirality from coords |
2D |
2D |
yes |
2D |
chirality from wedging |
3D |
2D |
yes |
2D |
chirality from wedging |
3D |
3D |
yes |
3D |
chirality from coords |
2D |
3D |
yes |
3D |
chirality from coords |
Here’s what the Molfile specification from Biovia says:
The “dimensional code” is maintained explicitly. Thus “3D” really means 3D, although “2D” will be interpreted as 3D if any non-zero Z-coordinates are found
We are consistent with this except for the case where the 3D flag is set for 2D coordinates and a wedge is present, in which case we ignore the 3D flag, mark the conformer as 2D, and set the stereochemistry based on the wedging
In cases where no 2D/3D flag is provided, the default value of the flag is 2D.
In a 2D structure, wedging is interpreted as a signal to indicate that stereochemistry is present and to indicate what the stereochemistry is.
When 3D coordinates are provided, stereochemistry is perceived from the coordinates themselves. If wedging is also present it will be ignored; the 3D signal is “stronger” than the wedging signal. The exception to this rule is atropisomeric bonds (see Atropisomeric Bonds), where the wedging simply indicates that atropisomerism should be perceived, but the direction of the wedge is ignored.
Here’s some code demonstrating that (and acting as a test):
>>> from rdkit import Chem
>>> from rdkit.Chem import rdDistGeom
# start with a molecule without defined chirality
>>> m = Chem.MolFromSmiles('FC(Cl)(Br)I')
>>> m3d = Chem.Mol(m)
>>> rdDistGeom.EmbedMolecule(m3d,randomSeed=0xa100f)
0
>>> mb_2d_2d = Chem.MolToMolBlock(m) # 2D flag, 2D coords
>>> t = Chem.MolFromMolBlock(mb_2d_2d)
>>> t.GetConformer().Is3D()
False
>>> t.GetAtomWithIdx(1).GetChiralTag()
rdkit.Chem.rdchem.ChiralType.CHI_UNSPECIFIED
>>> mb_3d_2d = mb_2d_2d.replace('2D','3D') # 3D flag, 2D coords
>>> t = Chem.MolFromMolBlock(mb_3d_2d)
>>> t.GetConformer().Is3D()
True
>>> t.GetAtomWithIdx(1).GetChiralTag()
rdkit.Chem.rdchem.ChiralType.CHI_UNSPECIFIED
>>> mb_3d_3d = Chem.MolToMolBlock(m3d) # 3D flag, 3D coords
>>> t = Chem.MolFromMolBlock(mb_3d_3d)
>>> t.GetConformer().Is3D()
True
>>> t.GetAtomWithIdx(1).GetChiralTag()
rdkit.Chem.rdchem.ChiralType.CHI_TETRAHEDRAL_CW
>>> mb_2d_3d = mb_3d_3d.replace('3D','2D') # 2D flag, 3D coords
>>> t = Chem.MolFromMolBlock(mb_3d_3d)
>>> t.GetConformer().Is3D()
True
>>> t.GetAtomWithIdx(1).GetChiralTag()
rdkit.Chem.rdchem.ChiralType.CHI_TETRAHEDRAL_CW
# Now do a molecule with defined chirality, this will have wedged bonds in the mol blocks
>>> m = Chem.MolFromSmiles('F[C@@](Cl)(Br)I')
>>> m3d = Chem.Mol(m)
>>> rdDistGeom.EmbedMolecule(m3d,randomSeed=0xa100f)
0
>>> mb_2d_2d = Chem.MolToMolBlock(m) # 2D flag, 2D coords
>>> t = Chem.MolFromMolBlock(mb_2d_2d)
>>> t.GetConformer().Is3D()
False
>>> t.GetAtomWithIdx(1).GetChiralTag()
rdkit.Chem.rdchem.ChiralType.CHI_TETRAHEDRAL_CW
>>> mb_3d_2d = mb_2d_2d.replace('2D','3D') # 3D flag, 2D coords
>>> t = Chem.MolFromMolBlock(mb_3d_2d)
>>> t.GetConformer().Is3D()
False
>>> t.GetAtomWithIdx(1).GetChiralTag()
rdkit.Chem.rdchem.ChiralType.CHI_TETRAHEDRAL_CW
>>> mb_3d_3d = Chem.MolToMolBlock(m3d) # 3D flag, 3D coords
>>> t = Chem.MolFromMolBlock(mb_3d_3d)
>>> t.GetConformer().Is3D()
True
>>> t.GetAtomWithIdx(1).GetChiralTag()
rdkit.Chem.rdchem.ChiralType.CHI_TETRAHEDRAL_CW
>>> mb_2d_3d = mb_3d_3d.replace('3D','2D') # 2D flag, 3D coords
>>> t = Chem.MolFromMolBlock(mb_3d_3d)
>>> t.GetConformer().Is3D()
True
>>> t.GetAtomWithIdx(1).GetChiralTag()
rdkit.Chem.rdchem.ChiralType.CHI_TETRAHEDRAL_CW
Ring Finding and SSSR¶
[Section taken from “Getting Started” document]
As others have ranted about with more energy and eloquence than I intend to, the definition of a molecule’s smallest set of smallest rings is not unique.
In some high symmetry molecules, a “true” SSSR will give results that are unappealing.
For example, the SSSR for cubane only contains 5 rings, even though there are “obviously” 6. This problem can be fixed by implementing a small (instead of smallest) set of smallest rings algorithm that returns symmetric results.
This is the approach that we took with the RDKit in rdkit.Chem.GetSymmSSSR().
Because it is sometimes useful to know the “true” SSSR rings, there is a rdkit.Chem.GetSSSR() function which returns this potentially non-unique set of rings.
For situations where you just care about knowing whether or not atoms/bonds are in rings, the RDKit provides the function
rdkit.Chem.rdmolops.FastFindRings(). This does a depth-first traversal of the molecule graph and identifies atoms and bonds that
are in rings.
Stereochemistry¶
Types of stereochemistry supported¶
The RDKit currently fully supports tetrahedral atomic stereochemistry and cis/trans stereochemistry at double bonds. There is partial support for non-tetrahedral stereochemistry, see the section Support for non-tetrahedral atomic stereochemistry.
Identification of potential stereoatoms/stereobonds¶
As of the 2020.09 release the RDKit has two different ways of identifying potential stereoatoms/stereobonds:
The legacy approach:
AssignStereochemistry(). This approach does a reasonable job of recognizing potential stereocenters, including some para-stereochemistry. It also has the side effect of assigning approximate CIP labels to the atoms/bonds (see below). This is currently the default algorithm.The new approach:
FindPotentialStereo(). The new approach is both more accurate (particularly for para-stereochemistry) and faster. It will become the default in a future RDKit version.
A concrete example of the accuracy improvements arising from the new algorithm:
|
|
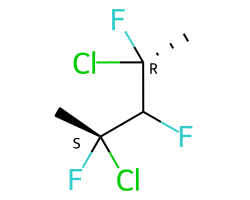
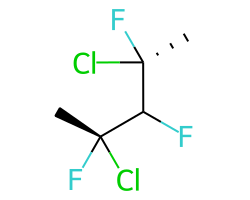
Both algorithms recognize that the central carbon is a potential stereocenter in the molecule on the left, but the old algorithm is unable to recognize it as a potential stereocenter in the molecule on the right.
Assignment of absolute stereochemistry¶
As of the 2020.09 release the RDKit has two different ways of assigning absolute stereochemistry labels (CIP labels):
The legacy approach uses an adaptation of an approximate algorithm for assigning CIP codes published by Paul Labute, [12]. The algorithm is reliable for determining whether or not a particular specified stereoatom/stereobond actually is a stereoatom/stereobond, but the CIP codes which it assigns are only truly correct for simple examples. As of the 2020.09 release this is the default algorithm, but this will be changed in a future RDKit release.
The new approach uses an implementation of a much more accurate algorithm, [13]. The new algorithm is more computationally expensive than the old one and does not provide CIP rankings of atoms (the concept of a global ranking of atoms isn’t well defined within the context of the true CIP algorithm). If you’re interested in having a chirality-sensitive ranking of all atoms, you can use the canonical atom ranking code instead.
Stereogenic atoms/bonds¶
The definitions of potential stereogenic atoms or bonds is inspired by the InChI definitions.
Stereogenic bonds¶
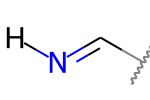
A double bond is potentially stereogenic if both atoms have at least two heavy atom neighbors and it’s not present in a ring with less than eight atoms.
For example, both of these double bonds are candidates for stereochemistry:
|
|
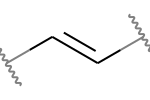
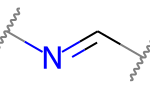
But this one is not:
Tetrahedral Stereogenic atoms¶
The following atom types are potential tetrahedral stereogenic atoms:
atoms with degree 4
atoms with degree 3 and one implicit H
P or As with degree 3 or 4
An SP3 hybridized N with degree 3 that is not involved in any conjugated bonds and that is in a ring of size 3 or that is shared between at least 3 rings (this last condition is an extension to the InChI rules).
S or Se with degree 3 and a total valence of 4 or a total valence of 3 and a net charge of +1.
Brief description of the findPotentialStereo() algorithm¶
Identify all potential stereogenic atoms and bonds in the molecule. If there aren’t any we don’t need to do anything else.
Foreach potential stereogenic atom: save the original chiral tag and then set the chiral tag to CW. Assign an atom symbol that makes this atom unique from all others (this will be used below in the canonicalization algorithm)
Foreach potential stereogenic bond: assign a bond symbol that makes this bond unique from all others (this will be used below in the canonicalization algorithm)
Determine the canonical atom ranking taking chirality into account, but not breaking ties. This uses the same canonicalization algorithm that’s used to generate SMILES. [14]
Remove the chiral tag from any potential stereogenic atom which has two identically ranked neighbors and set its symbol to the default for that atom
Set the symbol of any double bond which has two identically ranked atoms attached to either end [15] to the default for that bond
If steps 5 and 6 modfied any atoms or bonds, loop back to step 4.
Add any potential stereogenic atom which does not have to identically ranked neighbors to the results
Add any potential stereogenic atom which does not have two identically ranked atoms attached to either end [15] to the results
Return the results
Sources of information about stereochemistry¶
From SMILES¶
Atomic stereochemistry can be specified using @, @@, @SP, etc.
Potential stereocenters with no information provided are
ChiralType::CHI_UNSPECIFIED.
Double-bond stereochemistry is specfied using / and \ to indicate the
directionality of the neighboring single bonds. Double bonds with no stereo
information provided are BondStereo::STEREONONE.
From Mol¶
Atomic stereochemistry can be specified using wedged bonds if 2D coordinates are
present. If 3D coordinates are present, they are used to set the stereochemistry
for stereogenic atoms. Wiggly bonds (CFG=2 in V3000 mol blocks) set the
chiral tag of stereogenic start atom to ChiralType::CHI_UNSPECIFIED.
Double-bond stereochemistry is automatically set using the atomic coordinates;
this is true for both 2D and 3D coordinates. If a stereogenic double bound is
crossed (CFG=2 in V3000 mol blocks) or has an adjacent wiggly single bond
(CFG=2 in V3000 mol blocks), then it will be BondStereo::STEREOANY.
From CXSMILES¶
An initial stereochemistry assignment is done following the SMILES rules (see above).
A w: (wiggly bond) specification will set the stereochemistry of the start
atom to ChiralType::CHI_UNSPECIFIED and double bonds to
BondStereo::STEREOANY. Stereochemistry of ring bonds can be set using t,
c, or ctu.
If 2D coordinates are present in the CXSMILES, atomic stereo can be set using
`wU` or `wD` to create wedged bonds.
If 3D coordinates are present in the CXSMILES, they are used to set the
stereochemistry for stereogenic atoms and bonds. This supersedes other
specifications in the CXSMILES except for ctu and w.
Support for non-tetrahedral atomic stereochemistry¶
Starting with the 2022.09 release, the RDKit has partial, but evolving, support for non-tetrahedral stereochemistry. The status of this work is being tracked in this github issue: https://github.com/rdkit/rdkit/issues/4851
This code is being released in a preliminary state in order to get feedback as soon as we can and to start to gather experience working with these systems.
Status as of 2022.09.1 release¶
“Complete”¶
(Note that since is new territory, the term “complete” should be taken with a grain of salt.)
The basic representation
Parsing SMILES and SMARTS
Generation of 2D coordinates
Assignment of non-tetrahedral stereo from 3D structures
Partial¶
Writing SMILES. The SMILES generated should be correct, but they are not canonical.
Generation of 3D coordinates. The basics here work but the “chirality” of TBP and OH structures is not correct.
Writing mol files. Need wedged bonds for these to actually be done
Totally missing¶
Wedging bonds
Writing SMARTS
Substructure search integration
CIP assignment
Canonicalization
Stereochemistry cleanup: recognizing incorrect stereochemistry specifications
Assignment of non-tetrahedral stereo from 2D structures
SMILES notation¶
This discussion of the SMILES notation is drawn heavily from the OpenSMILES documentation: http://opensmiles.org/opensmiles.html Many thanks to the team which put that document together and to John Mayfield for his excellent CDK Depict tool, which I used double check my work on this.
The representation has a tag for what the stereo is, e.g. @SP, and a permutation number.
Square planar¶
@SP1 |
@SP2 |
@SP3 |
|---|---|---|
|
|
|
U |
4 |
Z |
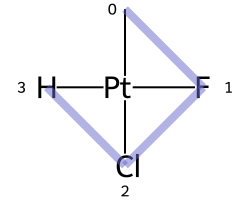
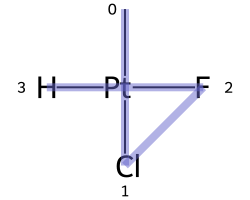
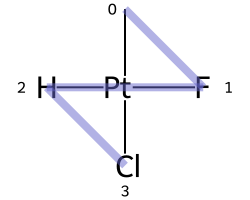
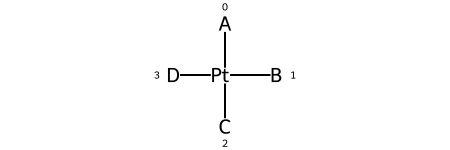
Here are the ligand numberings for the 3 possible permutations of the sample molecule:
Label |
A |
B |
C |
D |
SMILES |
|---|---|---|---|---|---|
@SP1 |
0 |
1 |
2 |
3 |
|
@SP2 |
0 |
2 |
1 |
3 |
|
@SP3 |
0 |
1 |
3 |
2 |
|
Trigonal bipyramidal¶
Here’s a specific example (from the OpenSMILES docs):
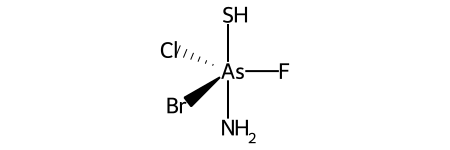
Here are the ligand labels and the ligand numbering for @TB1:
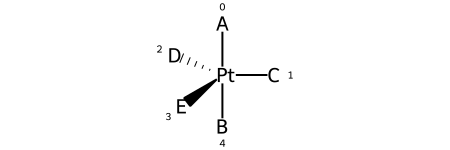
And then the ligand numberings for the 20 possible permutations of the sample molecule:
Label |
A |
B |
C |
D |
E |
SMILES |
|---|---|---|---|---|---|---|
@TB1 |
0 |
4 |
1 |
2 |
3 |
|
@TB2 |
0 |
4 |
1 |
3 |
2 |
|
@TB3 |
0 |
3 |
1 |
2 |
4 |
|
@TB4 |
0 |
3 |
1 |
4 |
2 |
|
@TB5 |
0 |
2 |
1 |
3 |
4 |
|
@TB6 |
0 |
2 |
1 |
4 |
3 |
|
@TB7 |
0 |
1 |
2 |
3 |
4 |
|
@TB8 |
0 |
1 |
2 |
4 |
3 |
|
@TB9 |
1 |
4 |
0 |
2 |
3 |
|
@TB11 |
1 |
4 |
0 |
3 |
2 |
|
@TB10 |
1 |
3 |
0 |
2 |
4 |
|
@TB12 |
1 |
3 |
0 |
4 |
2 |
|
@TB13 |
1 |
2 |
0 |
3 |
4 |
|
@TB14 |
1 |
2 |
0 |
4 |
3 |
|
@TB15 |
2 |
4 |
0 |
1 |
3 |
|
@TB20 |
2 |
4 |
0 |
3 |
1 |
|
@TB16 |
2 |
3 |
0 |
1 |
4 |
|
@TB19 |
2 |
3 |
0 |
4 |
1 |
|
@TB17 |
3 |
4 |
0 |
1 |
2 |
|
@TB18 |
3 |
4 |
0 |
2 |
1 |
|
Octahedral¶
Here’s a specific example (an invented molecule):
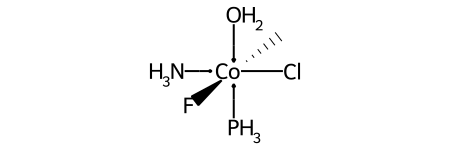
Here are the ligand labels and the ligand numbering for @OH1:
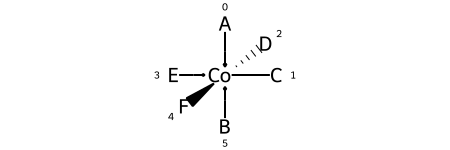
And then the square planar shape and ligand numberings for the 30 possible permutations of the sample molecule:
Label |
SP |
A |
B |
C |
D |
E |
F |
SMILES |
|---|---|---|---|---|---|---|---|---|
@OH1 |
U |
0 |
5 |
1 |
2 |
3 |
4 |
|
@OH2 |
U |
0 |
5 |
1 |
4 |
3 |
2 |
|
@OH3 |
U |
0 |
4 |
1 |
2 |
3 |
5 |
|
@OH16 |
U |
0 |
4 |
1 |
5 |
3 |
2 |
|
@OH6 |
U |
0 |
3 |
1 |
2 |
4 |
5 |
|
@OH18 |
U |
0 |
3 |
1 |
5 |
4 |
2 |
|
@OH19 |
U |
0 |
2 |
1 |
3 |
4 |
5 |
|
@OH24 |
U |
0 |
2 |
1 |
5 |
4 |
3 |
|
@OH25 |
U |
0 |
1 |
2 |
3 |
4 |
5 |
|
@OH30 |
U |
0 |
1 |
2 |
5 |
4 |
3 |
|
@OH4 |
Z |
0 |
5 |
1 |
2 |
4 |
3 |
|
@OH14 |
Z |
0 |
5 |
1 |
3 |
4 |
2 |
|
@OH5 |
Z |
0 |
4 |
1 |
2 |
5 |
3 |
|
@OH15 |
Z |
0 |
4 |
1 |
3 |
5 |
2 |
|
@OH7 |
Z |
0 |
3 |
1 |
2 |
5 |
4 |
|
@OH17 |
Z |
0 |
3 |
1 |
4 |
5 |
2 |
|
@OH20 |
Z |
0 |
2 |
1 |
3 |
5 |
4 |
|
@OH23 |
Z |
0 |
2 |
1 |
4 |
5 |
3 |
|
@OH26 |
Z |
0 |
1 |
2 |
3 |
5 |
4 |
|
@OH29 |
Z |
0 |
1 |
2 |
4 |
5 |
3 |
|
@OH10 |
4 |
0 |
5 |
1 |
4 |
2 |
3 |
|
@OH8 |
4 |
0 |
5 |
1 |
3 |
2 |
4 |
|
@OH11 |
4 |
0 |
4 |
1 |
5 |
2 |
3 |
|
@OH9 |
4 |
0 |
4 |
1 |
3 |
2 |
5 |
|
@OH13 |
4 |
0 |
3 |
1 |
4 |
2 |
4 |
|
@OH12 |
4 |
0 |
3 |
1 |
4 |
2 |
5 |
|
@OH22 |
4 |
0 |
2 |
1 |
5 |
3 |
4 |
|
@OH21 |
4 |
0 |
2 |
1 |
4 |
3 |
5 |
|
@OH28 |
4 |
0 |
1 |
2 |
5 |
3 |
4 |
|
@OH27 |
4 |
0 |
1 |
2 |
4 |
3 |
5 |
|
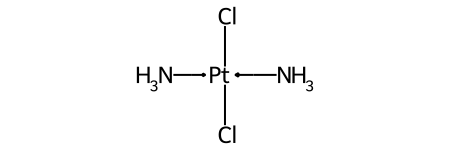
Duplicate ligands¶
One of the major differences between non-tetrahedral stereochemistry and the tetrahedral variant is that it’s possible to have non-tetrahedral stereo with central atoms which have duplicate ligands.
The classic example of this is cis-platin - Cl[Pt@SP1](Cl)(<-[NH3])<-[NH3] - vs trans-platin - Cl[Pt@SP2](Cl)(<-[NH3])<-[NH3] -
|
|
|---|---|
|
|
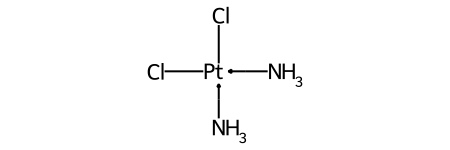
Treatment of implicit Hs¶
Implicit Hs are treated the same as in tetrahedral stereo: as if they are the
first neighbors after the central atom. So the two smiles C[Pt@SP1H](Cl)F
and C[Pt@SP1]([H])(Cl)F corresponds to the same structure.
This also works with multiple implicit Hs: C[Pt@SP1H2]Cl and C[Pt@SP1]([H])([H])Cl are equivalent.
Missing ligands¶
Coordination environments with missing ligands are treated as if the missing ligands were at the end of the ligand ordering.
For example, this invented complex can be presented with the SMILES O[Mn@OH1](Cl)(C)(N)F.
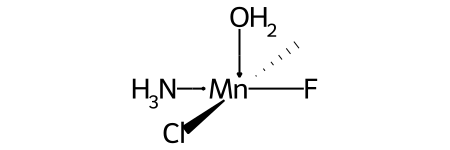
Compare this to the SMILES for the related complex shown above in the discussion of @OH stereo.
Chemical Reaction Handling¶
Reaction SMARTS¶
Not SMIRKS [1] , not reaction SMILES [2], derived from SMARTS [3].
The general grammar for a reaction SMARTS is :
reaction ::= reactants ">>" products
reactants ::= molecules
products ::= molecules
molecules ::= molecule
molecules "." molecule
molecule ::= a valid SMARTS string without "." characters
"(" a valid SMARTS string without "." characters ")"
Some features¶
Mapped dummy atoms in the product template are replaced by the corresponding atom in the reactant:
>>> from rdkit.Chem import AllChem
>>> rxn = AllChem.ReactionFromSmarts('[C:1]=[O,N:2]>>[C:1][*:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CC=O'),))[0]]
['CCO']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CC=N'),))[0]]
['CCN']
but unmapped dummy atoms are left as dummies:
>>> rxn = AllChem.ReactionFromSmarts('[C:1]=[O,N:2]>>*[C:1][*:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CC=O'),))[0]]
['*C(C)O']
“Any” bonds in the products are replaced by the corresponding bond in the reactant:
>>> rxn = AllChem.ReactionFromSmarts('[C:1]~[O,N:2]>>*[C:1]~[*:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('C=O'),))[0]]
['*C=O']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CO'),))[0]]
['*CO']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('C#N'),))[0]]
['*C#N']
Intramolecular reactions can be expressed flexibly by including reactants in parentheses. This is demonstrated in this ring-closing metathesis example [5]:
>>> rxn = AllChem.ReactionFromSmarts("([C:1]=[C;H2].[C:2]=[C;H2])>>[*:1]=[*:2]")
>>> m1 = Chem.MolFromSmiles('C=CCOCC=C')
>>> ps = rxn.RunReactants((m1,))
>>> Chem.MolToSmiles(ps[0][0])
'C1=CCOC1'
Chirality¶
This section describes how chirality information in the reaction defition is handled. A consistent example, esterification of secondary alcohols, is used throughout [6].
If no chiral information is present in the reaction definition, the stereochemistry of the reactants is preserved, as is membership in enhanced stereo groups:
>>> alcohol1 = Chem.MolFromSmiles('CC(CCN)O')
>>> alcohol2 = Chem.MolFromSmiles('C[C@H](CCN)O')
>>> alcohol3 = Chem.MolFromSmiles('C[C@@H](CCN)O')
>>> acid = Chem.MolFromSmiles('CC(=O)O')
>>> rxn = AllChem.ReactionFromSmarts('[CH1:1][OH:2].[OH][C:3]=[O:4]>>[C:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@@H](C)CCN'
You get the same result (retention of stereochemistry) if a mapped atom has the same chirality in both reactants and products:
>>> rxn = AllChem.ReactionFromSmarts('[C@H1:1][OH:2].[OH][C:3]=[O:4]>>[C@:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@@H](C)CCN'
A mapped atom with different chirality in reactants and products leads to inversion of stereochemistry:
>>> rxn = AllChem.ReactionFromSmarts('[C@H1:1][OH:2].[OH][C:3]=[O:4]>>[C@@:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
If a mapped atom has chirality specified in the reactants, but not in the products, the reaction destroys chirality at that center:
>>> rxn = AllChem.ReactionFromSmarts('[C@H1:1][OH:2].[OH][C:3]=[O:4]>>[C:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
And, finally, if chirality is specified in the products, but not the reactants, the reaction creates a stereocenter with the specified chirality:
>>> rxn = AllChem.ReactionFromSmarts('[CH1:1][OH:2].[OH][C:3]=[O:4]>>[C@:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
This doesn’t make sense without including a bit more context around the stereocenter in the reaction definition:
>>> rxn = AllChem.ReactionFromSmarts('[CH3:5][CH1:1]([C:6])[OH:2].[OH][C:3]=[O:4]>>[C:5][C@:1]([C:6])[O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
Note that the chirality specification is not being used as part of the query: a molecule with no chirality specified can match a reactant with specified chirality.
In general, the reaction machinery tries to preserve as much stereochemistry information as possible. This works when a single new bond is formed to a chiral center:
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C:2]-O>>[C:1][C:2]-S')
>>> alcohol2 = Chem.MolFromSmiles('C[C@@H](O)CCN')
>>> ps=rxn.RunReactants((alcohol2,))
>>> Chem.MolToSmiles(ps[0][0],True)
'C[C@@H](S)CCN'
But it fails if two or more bonds are formed:
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C:2](-O)-F>>[C:1][C:2](-S)-Cl')
>>> alcohol = Chem.MolFromSmiles('C[C@@H](O)F')
>>> ps=rxn.RunReactants((alcohol,))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(S)Cl'
In this case, there’s just not sufficient information present to allow the information to be preserved. You can help by providing mapping information:
Some caveats We made this code as robust as we can, but this is a non-trivial problem and it’s certainly possible to get surprising results.
Things get tricky if atom ordering around a chiral center changes in the reaction SMARTS. Here are some of the situations that are currently handled correctly.
Reordering of the neighbors, but the number and atom mappings of neighbors remains constant. In this case there is no inversion of chirality even though the chiral tag on the chiral atom changes between the reactants and products:
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C@:2]([F:3])[Br:4]>>[C:1][C@@:2]([S:4])[F:3]')
>>> mol = Chem.MolFromSmiles('C[C@@H](F)Br')
>>> ps=rxn.RunReactants((mol,))
>>> Chem.MolToSmiles(ps[0][0],True)
'C[C@@H](F)S'
Adding a neighbor to a chiral atom.
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C@H:2]([F:3])[Br:4]>>[C:1][C@@:2](O)([F:3])[Br:4]')
>>> mol = Chem.MolFromSmiles('C[C@@H](F)Br')
>>> ps=rxn.RunReactants((mol,))
>>> Chem.MolToSmiles(ps[0][0],True)
'C[C@](O)(F)Br'
Removing a neighbor from a chiral atom.
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C@:2](O)([F:3])[Br:4]>>[C:1][C@@H:2]([F:3])[Br:4]')
>>> mol = Chem.MolFromSmiles('C[C@@](O)(F)Br')
>>> ps=rxn.RunReactants((mol,))
>>> Chem.MolToSmiles(ps[0][0],True)
'C[C@H](F)Br'
Rules and warnings¶
Include atom map information at the end of an atom query. So do [C,N,O:1] or [C;R:1].
Don’t forget that unspecified bonds in SMARTS are either single or aromatic. Bond orders in product templates are assigned when the product template itself is constructed and it’s not always possible to tell if the bond should be single or aromatic:
>>> rxn = AllChem.ReactionFromSmarts('[#6:1][#7,#8:2]>>[#6:1][#6:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('C1NCCCC1'),))[0]]
['C1CCCCC1']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('c1ncccc1'),))[0]]
['c1ccccc-1']
So if you want to copy the bond order from the reactant, use an “Any” bond:
>>> rxn = AllChem.ReactionFromSmarts('[#6:1][#7,#8:2]>>[#6:1]~[#6:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('c1ncccc1'),))[0]]
['c1ccccc1']
The Feature Definition File Format¶
An FDef file contains all the information needed to define a set of chemical features. It contains definitions of feature types that are defined from queries built up using Daylight’s SMARTS language. [3] The FDef file can optionally also include definitions of atom types that are used to make feature definitions more readable.
Chemical Features¶
Chemical features are defined by a Feature Type and a Feature Family. The Feature Family is a general classification of the feature (such as “Hydrogen-bond Donor” or “Aromatic”) while the Feature Type provides additional, higher-resolution, information about features. Pharmacophore matching is done using Feature Family’s. Each feature type contains the following pieces of information:
A SMARTS pattern that describes atoms (one or more) matching the feature type.
Weights used to determine the feature’s position based on the positions of its defining atoms.
Syntax of the FDef file¶
AtomType definitions¶
An AtomType definition allows you to assign a shorthand name to be used in place of a SMARTS string defining an atom query. This allows FDef files to be made much more readable. For example, defining a non-polar carbon atom like this:
AtomType Carbon_NonPolar [C&!$(C=[O,N,P,S])&!$(C#N)]
creates a new name that can be used anywhere else in the FDef file that it would be useful to use this SMARTS. To reference an AtomType, just include its name in curly brackets. For example, this excerpt from an FDef file defines another atom type - Hphobe - which references the Carbon_NonPolar definition:
AtomType Carbon_NonPolar [C&!$(C=[O,N,P,S])&!$(C#N)]
AtomType Hphobe [{Carbon_NonPolar},c,s,S&H0&v2,F,Cl,Br,I]
Note that {Carbon_NonPolar} is used in the new AtomType definition without any additional decoration (no square brackes or recursive SMARTS markers are required).
Repeating an AtomType results in the two definitions being combined using the SMARTS “,” (or) operator. Here’s an example:
AtomType d1 [N&!H0]
AtomType d1 [O&!H0]
This is equivalent to:
AtomType d1 [N&!H0,O&!H0]
Which is equivalent to the more efficient:
AtomType d1 [N,O;!H0]
Note that these examples tend to use SMARTS’s high-precedence and operator “&” and not the low-precedence and “;”. This can be important when AtomTypes are combined or when they are repeated. The SMARTS “,” operator is higher precedence than “;”, so definitions that use “;” can lead to unexpected results.
It is also possible to define negative AtomType queries:
AtomType d1 [N,O,S]
AtomType !d1 [H0]
The negative query gets combined with the first to produce a definition identical to this:
AtomType d1 [!H0;N,O,S]
Note that the negative AtomType is added to the beginning of the query.
Feature definitions¶
A feature definition is more complex than an AtomType definition and stretches across multiple lines:
DefineFeature HDonor1 [N,O;!H0]
Family HBondDonor
Weights 1.0
EndFeature
The first line of the feature definition includes the feature type and the SMARTS string defining the feature. The next two lines (order not important) define the feature’s family and its atom weights (a comma-delimited list that is the same length as the number of atoms defining the feature). The atom weights are used to calculate the feature’s locations based on a weighted average of the positions of the atom defining the feature. More detail on this is provided below. The final line of a feature definition must be EndFeature. It is perfectly legal to mix AtomType definitions with feature definitions in the FDef file. The one rule is that AtomTypes must be defined before they are referenced.
Additional syntax notes:¶
Any line that begins with a # symbol is considered a comment and will be ignored.
A backslash character, , at the end of a line is a continuation character, it indicates that the data from that line is continued on the next line of the file. Blank space at the beginning of these additional lines is ignored. For example, this AtomType definition:
AtomType tButylAtom [$([C;!R](-[CH3])(-[CH3])(-[CH3])),\ $([CH3](-[C;!R](-[CH3])(-[CH3])))]
is exactly equivalent to this one:
AtomType tButylAtom [$([C;!R](-[CH3])(-[CH3])(-[CH3])),$([CH3](-[C;!R](-[CH3])(-[CH3])))]
(though the first form is much easier to read!)
Atom weights and feature locations¶
Frequently Asked Question(s)¶
What happens if a Feature Type is repeated in the file? Here’s an example:
DefineFeature HDonor1 [O&!H0] Family HBondDonor Weights 1.0 EndFeature DefineFeature HDonor1 [N&!H0] Family HBondDonor Weights 1.0 EndFeature
In this case both definitions of the HDonor1 feature type will be active. This is functionally identical to:
DefineFeature HDonor1 [O,N;!H0] Family HBondDonor Weights 1.0 EndFeature
However the formulation of this feature definition with a duplicated feature type is considerably less efficient and more confusing than the simpler combined definition.
Representation of Pharmacophore Fingerprints¶
In the RDKit scheme the bit ids in pharmacophore fingerprints are not hashed: each bit corresponds to a particular combination of features and distances. A given bit id can be converted back to the corresponding feature types and distances to allow interpretation. An illustration for 2D pharmacophores is shown in Figure 1: Bit numbering in pharmacophore fingerprints.

Figure 1: Bit numbering in pharmacophore fingerprints¶
Atom-Atom Matching in Substructure Queries¶
When doing substructure matches for queries derived from SMARTS the rules for which atoms in the molecule should match which atoms in the query are well defined.[#smarts]_ The same is not necessarily the case when the query molecule is derived from a mol block or SMILES.
The general rule used in the RDKit is that if you don’t specify a property in the query, then it’s not used as part of the matching criteria and that Hs are ignored. This leads to the following behavior:
Molecule |
Query |
Match |
|---|---|---|
CCO |
CCO |
Yes |
CC[O-] |
CCO |
Yes |
CCO |
CC[O-] |
No |
CC[O-] |
CC[O-] |
Yes |
CC[O-] |
CC[OH] |
Yes |
CCOC |
CC[OH] |
Yes |
CCOC |
CCO |
Yes |
CCC |
CCC |
Yes |
CC[14C] |
CCC |
Yes |
CCC |
CC[14C] |
No |
CC[14C] |
CC[14C] |
Yes |
OCO |
C |
Yes |
OCO |
[CH] |
No |
OCO |
[CH2] |
No |
OCO |
[CH3] |
No |
OCO |
O[CH3] |
Yes |
O[CH2]O |
C |
Yes |
O[CH2]O |
[CH2] |
No |
Demonstrated here:
>>> Chem.MolFromSmiles('CCO').HasSubstructMatch(Chem.MolFromSmiles('CCO'))
True
>>> Chem.MolFromSmiles('CC[O-]').HasSubstructMatch(Chem.MolFromSmiles('CCO'))
True
>>> Chem.MolFromSmiles('CCO').HasSubstructMatch(Chem.MolFromSmiles('CC[O-]'))
False
>>> Chem.MolFromSmiles('CC[O-]').HasSubstructMatch(Chem.MolFromSmiles('CC[O-]'))
True
>>> Chem.MolFromSmiles('CC[O-]').HasSubstructMatch(Chem.MolFromSmiles('CC[OH]'))
True
>>> Chem.MolFromSmiles('CCOC').HasSubstructMatch(Chem.MolFromSmiles('CC[OH]'))
True
>>> Chem.MolFromSmiles('CCOC').HasSubstructMatch(Chem.MolFromSmiles('CCO'))
True
>>> Chem.MolFromSmiles('CCC').HasSubstructMatch(Chem.MolFromSmiles('CCC'))
True
>>> Chem.MolFromSmiles('CC[14C]').HasSubstructMatch(Chem.MolFromSmiles('CCC'))
True
>>> Chem.MolFromSmiles('CCC').HasSubstructMatch(Chem.MolFromSmiles('CC[14C]'))
False
>>> Chem.MolFromSmiles('CC[14C]').HasSubstructMatch(Chem.MolFromSmiles('CC[14C]'))
True
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('C'))
True
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('[CH]'))
False
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('[CH2]'))
False
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('[CH3]'))
False
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('O[CH3]'))
True
>>> Chem.MolFromSmiles('O[CH2]O').HasSubstructMatch(Chem.MolFromSmiles('C'))
True
>>> Chem.MolFromSmiles('O[CH2]O').HasSubstructMatch(Chem.MolFromSmiles('[CH2]'))
False
Generic (“Markush”) queries in substructure matching¶
Note This section describes functionality added in the 2022.03.1 release of the RDKit.
The RDKit supports a set of generic queries used as part of the Beilstein and Reaxys systems. Here’s an example:
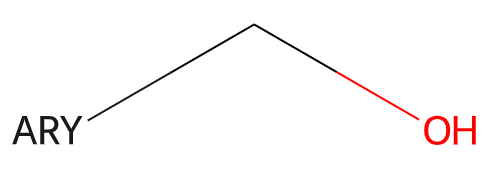
Information about generic queries can be read in from CXSMILES or V3000 Mol blocks (as SUP SGroups) and then calling the function Chem.SetGenericQueriesFromProperties() with the molecule to be modified as an argument. These features are not used by default when doing substructure queries, but can be enabled by setting the option SubstructMatchParameters.useGenericMatchers to True
Here’s an example of using the features:
>>> q = Chem.MolFromSmarts('OC* |$;;ARY$|')
>>> Chem.SetGenericQueriesFromProperties(q)
>>> Chem.MolFromSmiles('C1CCCCC1CO').HasSubstructMatch(q)
True
>>> Chem.MolFromSmiles('c1ccccc1CO').HasSubstructMatch(q)
True
>>> ps = Chem.SubstructMatchParameters()
>>> ps.useGenericMatchers = True
>>> Chem.MolFromSmiles('C1CCCCC1CO').HasSubstructMatch(q,ps)
False
>>> Chem.MolFromSmiles('c1ccccc1CO').HasSubstructMatch(q,ps)
True
Here are the supported groups and a brief description of what they mean:
Alkyl (ALK)
alkyl side chains (not an H atom)
AlkylH (ALH)
alkyl side chains including an H atom
Alkenyl (AEL)
alkenyl side chains
AlkenylH (AEH)
alkenyl side chains or an H atom
Alkynyl (AYL)
alkynyl side chains
AlkynylH (AYH)
alkynyl side chains or an H atom
Alkoxy (AOX)
alkoxy side chains
AlkoxyH (AOH)
alkoxy side chains or an H atom
Carbocyclic (CBC)
carbocyclic side chains
CarbocyclicH (CBH)
carbocyclic side chains or an H atom
Carbocycloalkyl (CAL)
cycloalkyl side chains
CarbocycloalkylH (CAH)
cycloalkyl side chains or an H atom
Carbocycloalkenyl (CEL)
cycloalkenyl side chains
CarbocycloalkenylH (CEH)
cycloalkenyl side chains or an H atom
Carboaryl (ARY)
all-carbon aryl side chains
CarboarylH (ARH)
all-carbon aryl side chains or an H atom
Cyclic (CYC)
cyclic side chains
CyclicH (CYH)
cyclic side chains or an H atom
Acyclic(ACY)
acyclic side chains (not an H atom)
AcyclicH (ACH)
acyclic side chains or an H atom
Carboacyclic (ABC)
all-carbon acyclic side chains
CarboacyclicH (ABH)
all-carbon acyclic side chains or an H atom
Heteroacyclic (AHC)
acyclic side chains with at least one heteroatom
HeteroacyclicH (AHH)
acyclic side chains with at least one heteroatom or an H atom
Heterocyclic (CHC)
cyclic side chains with at least one heteroatom
HeterocyclicH (CHH)
cyclic side chains with at least one heteroatom or an H atom
Heteroaryl (HAR)
aryl side chains with at least one heteroatom
HeteroarylH (HAH)
aryl side chains with at least one heteroatom or an H atom
NoCarbonRing (CXX)
ring containing no carbon atoms
NoCarbonRingH (CXH)
ring containing no carbon atoms or an H atom
Group (G)
any group (not H atom)
GroupH (GH)
any group (including H atom)
Group* (G*)
any group with a ring closure
GroupH* (GH*)
any group with a ring closure or an H atom
For more detailed descriptions, look at the documentation for the C++ file GenericGroups.h
Molecular Sanitization¶
The molecule parsing functions all, by default, perform a “sanitization” operation on the molecules read. The idea is to generate useful computed properties (like hybridization, ring membership, etc.) for the rest of the code and to ensure that the molecules are “reasonable”: that they can be represented with octet-complete Lewis dot structures.
Here are the steps involved, in order.
clearComputedProps: removes any computed properties that already existon the molecule and its atoms and bonds. This step is always performed.
cleanUp: standardizes a small number of non-standard valence states. The clean up operations are:
Neutral 5 valent Ns with double bonds to Os are converted to the zwitterionic form. Example:
N(=O)=O -> [N+](=O)O-]Neutral 5 valent Ns with triple bonds to another N are converted to the zwitterionic form. Example:
C-N=N#N -> C-N=[N+]=[N-]Neutral 5 valent phosphorus with one double bond to an O and another to either a C or a P are converted to the zwitterionic form. Example:
C=P(=O)O -> C=[P+]([O-])ONeutral Cl, Br, or I with exclusively O neighbors, and a valence of 3, 5, or 7, are converted to the zwitterionic form. This covers things like chlorous acid, chloric acid, and perchloric acid. Example:
O=Cl(=O)O -> [O-][Cl+2][O-]OThis step should not generate exceptions.
cleanUpOrganometallics: standardizes a small number of non-standard situations encountered in organometallics. The cleanup operations are:
replaces single bonds from hypervalent atoms to metals with dative bonds.
This step should not generate exceptions.
updatePropertyCache: calculates the explicit and implicit valences on all atoms. This generates exceptions for atoms in higher-than-allowed valence states. This step is always performed, but if it is “skipped” the test for non-standard valences will not be carried out.
symmetrizeSSSR: calls the symmetrized smallest set of smallest rings algorithm (discussed in the Getting Started document).
Kekulize: converts aromatic rings to their Kekule form. Will raise an exception if a ring cannot be kekulized or if aromatic bonds are found outside of rings.
assignRadicals: determines the number of radical electrons (if any) on each atom.
setAromaticity: identifies the aromatic rings and ring systems (see above), sets the aromatic flag on atoms and bonds, sets bond orders to aromatic.
setConjugation: identifies which bonds are conjugated
setHybridization: calculates the hybridization state of each atom
cleanupChirality: removes chiral tags from atoms that are not sp3 hybridized.
adjustHs: adds explicit Hs where necessary to preserve the chemistry. This is typically needed for heteroatoms in aromatic rings. The classic example is the nitrogen atom in pyrrole.
updatePropertyCache: re-calculates the explicit and implicit valences on all atoms. This generates exceptions for atoms in higher-than-allowed valence states. This step is required to catch some edge cases where input atoms with non-physical valences are accepted if they are flagged as aromatic.
The individual steps can be toggled on or off when calling
MolOps::sanitizeMol or Chem.SanitizeMol.
Valence calculation and allowed valences¶
The RDKit is, by default, fairly strict in the way it enforces allowed valences when sanitizing structures (this is done during the updatePropertyCache step of sanitization): atoms which have an explicit valence (sum of the specified bond orders + specified H count) exceeding the maximum allowed valence for the element will raise an exception.
Allowed valences of the elements (as of 2024.09.1):
H 1
He 0
Li 1 -1
Be 2
B 3
C 4
N 3
O 2
F 1
Ne 0
Na 1,-1
Mg 2,-1
Al 3
Si 4
P 3,5
S 2,4,6
Cl 1
Ar 0
K 1,-1
Ca 2,-1
Ga 3
Ge 4
As 3,5
Se 2,4,6
Br 1
Kr 0
Rb 1,-1
Sr 2,-1
In 3
Sn 2,4
Sb 3,5
Te 2,4,6
I 1,3,5
Xe 0,2,4,6
Cs 1,-1
Ba 2,-1
Tl -1
Pb 2,4
Bi 3,5
Po 2,4,6
At 1,3,5
Rn 0
Elements not listed in the table have a valence of -1.
An allowed valence of -1 indicates that the element can have any valence value. Implicit Hs will not be added to atoms with a possible valence of -1 when the explicit valence exceeds the highest specified value. So, for example, an Mg atom with a single bond to it (explicit valence = 1) will have one implicit H added to it, while an Mg atom with three bonds to it will have no implicit Hs added. Atoms where the only allowed valence is -1 will never have implicit Hs added.
The allowed valences of charged atoms are calculated by looking at the isoelectronic element’s allowed valences. For example, N+ has the same allowed valences as C, while N- has the same allowed valences as O. P-2, S-, As-2, and Se- are special cases: they all have allowed valences of 1, 3 and 5.
JSON Support¶
The RDKit supports writing to/reading from two closely related JSON formats: commonchem (https://github.com/CommonChem/CommonChem) and rdkitjson. commonchem is a well-documented format designed to be used for efficient interchange between molecular toolkits. rdkitjson is an extension to commonchem which includes additional features allowing RDKit molecules to be serialized to JSON. The extensions in rdkitjson - enhanced stereo and substance groups - are generally useful, so it’s easy to imagine them being integrated into commonchem at some point in the future.
Lists of molecules can be converted to JSON with MolInterchange::MolsToJSONData() (C++) or Chem.MolsToJSONData() (Python). Those calls take an optional parameters object which can be used to specify whether commonchem or rdkitjson is generated. The default is to generate rdkitjson.
JSON data can be converted back to RDKit molecules using MolInterchange::JSONDataToMols() (C++) or Chem.JSONDataToMols() (Python). The parser will automatically determine whether or not its working with commonchem or rdkitjson.
rdkitjson format¶
Enhanced stereo¶
Here’s the rdkitjson representation of the stereo groups from the molecule C[C@@H]1C([C@H](O)F)O[C@H](C)C([C@@H](O)F)[C@@H]1C |a:7,o1:3,10,&1:1,&2:13|:
'stereoGroups': [{'type': 'abs', 'atoms': [7]},
{'type': 'or', 'atoms': [3, 10]},
{'type': 'and', 'atoms': [1]},
{'type': 'and', 'atoms': [13]}],
Substance groups¶
Here’s the rdkitjson representation of a SUP substance group:
'substanceGroups': [{'properties': {'TYPE': 'SUP',
'index': 1,
'LABEL': 'Boc',
'DATAFIELDS': '[]'},
'atoms': [7, 8, 9, 10, 11, 12, 13],
'bonds': [8],
'brackets': [[[6.24, -2.9, 0.0], [6.24, -2.9, 0.0], [0.0, 0.0, 0.0]]],
'cstates': [{'bond': 8, 'vector': [0.0, 0.82, 0.0]}],
'attachPoints': [{'aIdx': 12, 'lvIdx': 5, 'id': '1'}]}],
and one for an SRU group:
'substanceGroups': [{'properties': {'TYPE': 'SRU',
'index': 1,
'CONNECT': 'HT',
'LABEL': 'n',
'DATAFIELDS': '[]'},
'atoms': [2, 1, 4],
'bonds': [2, 0],
'brackets': [[[-3.9538, 4.3256, 0.0],
[-3.0298, 2.7252, 0.0],
[0.0, 0.0, 0.0]],
[[-5.4618, 2.8611, 0.0],
[-6.3858, 4.4615, 0.0],
[0.0, 0.0, 0.0]]]}],
Implementation Details¶
“Magic” Property Values¶
The following property values are regularly used in the RDKit codebase and may be useful to client code.
ROMol (Mol in Python)¶
Property Name |
Use |
|---|---|
MolFileComments |
Read from/written to the comment line of CTABs. |
MolFileInfo |
Read from/written to the info line of CTABs. |
_MolFileChiralFlag |
Read from/written to the chiral flag of CTABs. |
_Name |
Read from/written to the name line of CTABs. |
_smilesAtomOutputOrder |
The order in which atoms were written to SMILES |
_smilesBondOutputOrder |
The order in which bonds were written to SMILES |
Atom¶
Property Name |
Use |
|---|---|
_CIPCode |
the CIP code (R or S) of the atom |
_CIPRank |
the integer CIP rank of the atom |
_ChiralityPossible |
set if an atom is a possible chiral center |
_MolFileRLabel |
integer R group label for an atom, read from/written to CTABs. |
_ReactionDegreeChanged |
set on an atom in a product template of a reaction if its degree changes in the reaction |
_protected |
atoms with this property set will not be considered as matching reactant queries in reactions |
dummyLabel |
(on dummy atoms) read from/written to CTABs as the atom symbol |
molAtomMapNumber |
the atom map number for an atom, read from/written to SMILES and CTABs |
molfileAlias |
the mol file alias for an atom (follows A tags), read from/written to CTABs |
molFileValue |
the mol file value for an atom (follows V tags), read from/written to CTABs |
molFileInversionFlag |
used to flag whether stereochemistry at an atom changes in a reaction, read from/written to CTABs, determined automatically from SMILES |
molRxnComponent |
which component of a reaction an atom belongs to, read from/written to CTABs |
molRxnRole |
which role an atom plays in a reaction (1=Reactant, 2=Product, 3=Agent), read from/written to CTABs |
smilesSymbol |
determines the symbol that will be written to a SMILES for the atom |
Thread safety and the RDKit¶
While writing the RDKit, we did attempt to ensure that the code would work in a multi-threaded environment by avoiding use of global variables, etc. However, making code thread safe is not a completely trivial thing, so there are no doubt some gaps. This section describes which pieces of the code base have explicitly been tested for thread safety.
- Note: With the exception of the small number of methods/functions
that take a
numThreadsargument, this section does not apply to using the RDKit from Python threads. Boost.Python ensures that only one thread is calling into the C++ code at any point. To get concurrent execution in Python, use the multiprocessing module or one of the other standard python approaches for this .
What has been tested¶
Reading molecules from SMILES/SMARTS/Mol blocks
Writing molecules to SMILES/SMARTS/Mol blocks (see below)
Generating 2D coordinates
Generating 3D conformations with the distance geometry code
Optimizing molecules with UFF or MMFF
Generating fingerprints
The descriptor calculators in $RDBASE/Code/GraphMol/Descriptors
Substructure searching (Note: if a query molecule contains recursive queries, it may not be safe to use it concurrently on multiple threads, see below)
The Subgraph code
The ChemTransforms code
The chemical reactions code
The Open3DAlign code
The MolDraw2D drawing code
The InChI code, with InChI IUPAC v1.06
Known Problems¶
The MolSuppliers (e.g. SDMolSupplier, SmilesMolSupplier?) change their internal state when a molecule is read. It is not safe to use one supplier on more than one thread.
Substructure searching using query molecules that include recursive queries. The recursive queries modify their internal state when a search is run, so it’s not safe to use the same query concurrently on multiple threads. If the code is built using the
RDK_BUILD_THREADSAFE_SSSargument (the default for the binaries we provide), a mutex is used to ensure that only one thread is using a given recursive query at a time.Calling MolToSmiles() on the same molecule from multiple threads can lead to data races with the calculated properties on the molecule.
Implementation of the TPSA Descriptor¶
The topological polar surface area (TPSA) descriptor implemented in the RDKit is described in a publication by Peter Ertl et al. (https://pubs.acs.org/doi/abs/10.1021/jm000942e) The RDKit’s implementation differs from what is described in that publication. This section describes the difference and why it’s there.
The RDKit’s TPSA implementation only includes, by default, contributions from N and O atoms. Table 1 of the TPSA publication. however, includes parameters for polar S and P in addition to N and O. What’s going on?
The original TPSA implementation that is in the Daylight Contrib dir (http://www.daylight.com/download/contrib/tpsa.html) does not include contributions from polar S or P and, it turns out, the reference values that are included in the TPSA paper also don’t include S or P contributions. For example, the TPSA provided in Table 3 for foscarnet (SMILES OC(=O)P(=O)(O)O), 94.8, corresponds the sum of the O contributions - 3x20.23 + 2*17.07 = 94.8. Adding the P contribution - 9.81- would give a PSA of 104.6. This is also true for the other P and S containing compounds in Table 3.
In the RDKit implementation, we chose to reproduce the behavior of the tpsa.c Contrib program and what is provided in Table 3 of the paper, so polar S and P are ignored. Based on a couple of user requests, for the 2018.09 release of the RDKit we added the option to include S and P contributions:
>>> from rdkit.Chem import Descriptors
>>> Descriptors.TPSA(Chem.MolFromSmiles('OC(=O)P(=O)(O)O')) # foscarnet
94.83
>>> Descriptors.TPSA(Chem.MolFromSmiles('OC(=O)P(=O)(O)O'), includeSandP=True)
104.64...
>>> Descriptors.TPSA(Chem.MolFromSmiles('Cc1ccccc1N1C(=O)c2cc(S(N)(=O)=O)c(Cl)cc2NC1C')) # metolazone
92.5
>>> Descriptors.TPSA(Chem.MolFromSmiles('Cc1ccccc1N1C(=O)c2cc(S(N)(=O)=O)c(Cl)cc2NC1C'), includeSandP=True)
100.88
Atom Properties and SDF files¶
Note This section describes functionality added in the 2019.03.1 release of the RDKit.
By default the rdkit.Chem.rdmolfiles.SDMolSupplier and rdkit.Chem.rdmolfiles.ForwardSDMolSupplier classes
(RDKit::SDMolSupplier and RDKit::ForwardMolSupplier in C++) can now recognize some molecular properties as property lists
and them into atomic properties. Properties with names that start with atom.prop, atom.iprop, atom.dprop, or atom.bprop
are converted to atomic properties of type string, int (64 bit), double, or bool respectively.
Here’s a sample block from an SDF that demonstrates all of the features, they are explained below:
property_example
RDKit 2D
3 3 0 0 0 0 0 0 0 0999 V2000
0.8660 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.4330 0.7500 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.4330 -0.7500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
2 3 1 0
3 1 1 0
M END
> <atom.dprop.PartialCharge> (1)
0.008 -0.314 0.008
> <atom.iprop.NumHeavyNeighbors> (1)
2 2 2
> <atom.prop.AtomLabel> (1)
C1 N2 C3
> <atom.bprop.IsCarbon> (1)
1 0 1
> <atom.prop.PartiallyMissing> (1)
one n/a three
> <atom.iprop.PartiallyMissingInt> (1)
[?] 2 2 ?
$$$$
Every atom property list should contain a number of space-delimited elements equal to the number of atoms.
Missing values are, by default, indicated with the string n/a. The missing value marker can be changed by beginning
the property list with a value in square brackets. So, for example, the property PartiallyMissing is set to “one”
for atom 0, “three” for atom 2, and is not set for atom 1. Similarly the property PartiallyMissingInt is set to 2 for atom 0, 2 for atom 1,
and is not set for atom 2.
This behavior is enabled by default and can be turned on/off with the
rdkit.Chem.rdmolfiles.SetProcessPropertyLists method.
If you have atom properties that you would like to have written to SDF files, you can use the functions
rdkit.Chem.rdmolfiles.CreateAtomStringPropertyList(), rdkit.Chem.rdmolfiles.CreateAtomIntPropertyList(),
rdkit.Chem.rdmolfiles.CreateAtomDoublePropertyList(), or rdkit.Chem.rdmolfiles.CreateAtomBoolPropertyList() :
>>> m = Chem.MolFromSmiles('CO')
>>> m.GetAtomWithIdx(0).SetDoubleProp('foo',3.14)
>>> Chem.CreateAtomDoublePropertyList(m,'foo')
>>> m.GetProp('atom.dprop.foo')
'3.1400000000000001 n/a'
>>> from io import StringIO
>>> sio = StringIO()
>>> w = Chem.SDWriter(sio)
>>> w.write(m)
>>> w=None
>>> print(sio.getvalue())
RDKit 2D
2 1 0 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2990 0.7500 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
M END
> <atom.dprop.foo> (1)
3.1400000000000001 n/a
$$$$
Support for Enhanced Stereochemistry¶
Overview¶
Enhanced stereochemistry is used to indicate that a molecule represents more than one possible diastereomer.
AND indicates that a molecule is a mixture of molecules. OR indicates unknown single substances,
and ABS indicates a single substance. This follows, the convention used in V3k mol files: groups of
atoms with specified stereochemistry with an ABS, AND, or OR marker indicating what is known.
Here are some illustrations of what the various combinations mean:
What’s drawn |
Mixture? |
What it means |
|---|---|---|
|
mixture |
|
|
mixture |
|
|
mixture |
|
|
single |
|
|
single |
|
|
single |
|
|
mixture |
|
|
single |
|
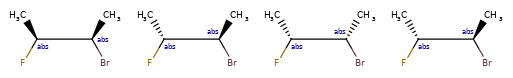
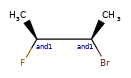
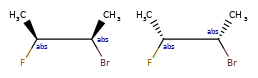
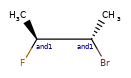
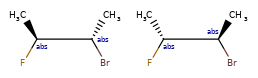
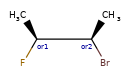
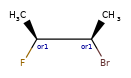
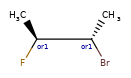
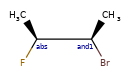
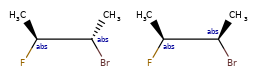
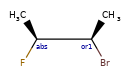
Representation¶
Stored as a vector of rdkit.Chem.rdchem.StereoGroup objects on a molecule. Each StereoGroup keeps track of its type
and the set of atoms that make it up.
Use cases¶
The initial target is to not lose data on an V3k mol -> RDKit -> V3k mol round trip. Manipulation and depiction are future goals.
It is possible to enumerate the elements of a StereoGroup using the function
rdkit.Chem.EnumerateStereoisomers.EumerateStereoisomers(). Note that
this removes the StereoGroup information from the products since they now
correspond to specific molecules:
>>> m = Chem.MolFromSmiles('C[C@H](F)C[C@H](O)Cl |a:4,&1:1|')
>>> m.GetStereoGroups()[0].GetGroupType()
rdkit.Chem.rdchem.StereoGroupType.STEREO_ABSOLUTE
>>> [x.GetIdx() for x in m.GetStereoGroups()[0].GetAtoms()]
[4]
>>> m.GetStereoGroups()[1].GetGroupType()
rdkit.Chem.rdchem.StereoGroupType.STEREO_AND
>>> [x.GetIdx() for x in m.GetStereoGroups()[1].GetAtoms()]
[1]
>>> from rdkit.Chem.EnumerateStereoisomers import EnumerateStereoisomers
>>> [Chem.MolToCXSmiles(x) for x in EnumerateStereoisomers(m)]
['C[C@@H](F)C[C@H](O)Cl', 'C[C@H](F)C[C@H](O)Cl']
Reactions also preserve StereoGroup``s. Product atoms are included in the ``StereoGroup as long as the reaction doesn’t create or destroy chirality at the atom.
>>> def clearAllAtomProps(mol):
... """So that atom mapping isn't shown"""
... for atom in mol.GetAtoms():
... for key in atom.GetPropsAsDict():
... atom.ClearProp(key)
...
>>> rxn = AllChem.ReactionFromSmarts('[C:1]F >> [C:1]Br')
>>> ps=rxn.RunReactants([m])
>>> clearAllAtomProps(ps[0][0])
>>> Chem.MolToCXSmiles(ps[0][0])
'C[C@H](Br)C[C@H](O)Cl |a:4,&1:1|'
Stereo Groups can be canonicalized.
>>> m = Chem.MolFromSmiles('CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@@H](C)CC1 |a:3,o1:11,o2:14|')
>>> mOut = Chem.CanonicalizeStereoGroups(m, Chem.StereoGroupAbsOptions.NeverInclude)
>>> Chem.MolToCXSmiles(mOut)
'CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@H](C)CC1 |o1:14|'
>>> mOut = Chem.CanonicalizeStereoGroups(m, Chem.StereoGroupAbsOptions.AlwaysInclude)
>>> Chem.MolToCXSmiles(mOut)
'CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@H](C)CC1 |a:3,11,o1:14|'
>>> mOut = Chem.CanonicalizeStereoGroups(m, Chem.StereoGroupAbsOptions.OnlyIncludeWhenOtherGroupsExist)
>>> Chem.MolToCXSmiles(mOut)
'CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@H](C)CC1 |a:3,11,o1:14|'
>>> m = Chem.MolFromSmiles('CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@@H](C)CC1 |a:3|')
>>> mOut = Chem.CanonicalizeStereoGroups(m, Chem.StereoGroupAbsOptions.OnlyIncludeWhenOtherGroupsExist)
>>> Chem.MolToCXSmiles(mOut)
'CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@@H](C)CC1'
Enhanced Stereochemistry and substructure search¶
Enhanced Stereochemistry may optionally be honored in substructure searches. The following table captures whether or not a substructure query (in the rows) matches a particular molecule (in the columns).
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|
|
Y |
Y |
Y |
Y |
Y |
Y |
Y |
|
N |
Y |
N |
N |
N |
Y |
Y |
|
N |
N |
Y |
N |
N |
Y |
Y |
|
N |
N |
N |
Y |
N |
N |
N |
|
N |
Y |
N |
N |
Y |
Y |
Y |
|
N |
N |
N |
N |
N |
Y |
Y |
|
N |
N |
N |
N |
N |
N |
Y |
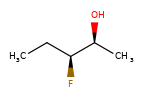
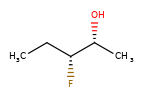
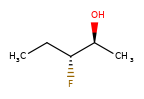
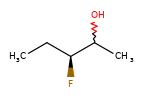
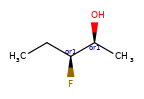
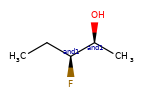
Substructure search using molecules with enhanced stereochemistry follows these rules (where substructure < superstructure):
achiral < everything, because an achiral query means ignore chirality in the match
chiral < AND, because AND includes both the chiral molecule and another one
chiral < OR, because OR includes either the chiral molecule or another one
OR < AND, because AND includes both molecules that OR could actually mean.
one group of two atoms < two groups of one atom, because the latter is 4 different diastereomers, and the former only two of the four.
Some concrete examples of this:
>>> ps = Chem.SubstructMatchParameters()
>>> ps.useChirality = True
>>> ps.useEnhancedStereo = True
>>> m_ABS = Chem.MolFromSmiles('CC[C@H](F)[C@H](C)O')
>>> m_AND = Chem.MolFromSmiles('CC[C@H](F)[C@H](C)O |&1:2,4|')
>>> m_OR = Chem.MolFromSmiles('CC[C@H](F)[C@H](C)O |o1:2,4|')
>>> m_AND.HasSubstructMatch(m_ABS,ps)
True
>>> m_OR.HasSubstructMatch(m_ABS,ps)
True
>>> m_AND.HasSubstructMatch(m_OR,ps)
True
>>> m_OR.HasSubstructMatch(m_AND,ps)
False
Atropisomeric Bonds¶
Some single bonds have restricted rotation because of steric interactions between the groups on adjacent atoms. If the groups on the adjacent atoms are different from each other, chirality can be induced. An atropisomer bond is such a restricted rotation bond.
The requirements for a bond to be eligible for atropisomerism in the RDKit are:
The bond must be a single bond between SP2 hybridized atoms.
The neighboring bonds must be single, double or aromatic.
If there are two groups on either end, those groups must be different as per CIP rules.
Currently RDKit considers ring bonds as potential atropisomer bonds only if the ring in which the bond appears is 8 atoms or larger (thus allowing macrocycles).
The molecule must have coordinates for atropisomer bonds to be interpreted.
The definition of potential atropisomer bonds is based on the wedging of adjacent bonds.
Defining Atropisomers¶
At least one of the neighbor bonds of one of the atoms of the potential atropisomer bond must be a single or aromatic bond, and must have a bond direction that is either “wedged” or “hashed”. If any of the neighbor bonds is marked as “sqwiggly”, the bond is considered to have “Any” stereochemistry. Example structure:
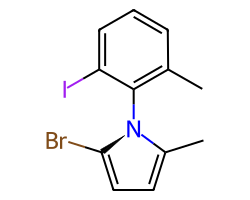
If more than one of the neighbor bonds are wedged or hashed, they must be consistent.
For example, if two neighbor bonds on the same end atom are wedged/hashed, one must be a wedge and the other must be a hash. If neighbor bonds on different ends of the atropisomer bond are wedged, and the two bonds are opposite sides of the potential atropisomer bond (the dihedral angle is greater than 90 degrees or less than -90 degrees), the two must both be wedges or both must be hashed. If the two wedged/hashed neighbor bonds are on the same side of the potential atropisomer bond (the dihedral angle is less than 90 degrees and greater than -90 degrees), one must be a wedge and the other a hash.
Examples – valid atropisomers with multiple wedges:
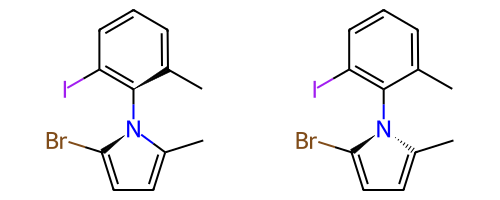
Examples – invalid atropisomers with multiple wedges:
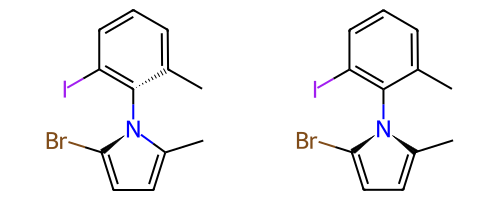
Note: the RDKit software makes no attempt to determine if the bond is actually rotationally constrained. If the bond meets the requirements above, it is marked as an atropisomer.
Internal Representation of Atropisomers¶
To help with the rest of the explanation, we include the bond indices in the molecule drawing:
Here’s part of the Debug output for that molecule:
Atoms:
...
1 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
...
5 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
...
7 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
8 7 N chg: 0 deg: 3 exp: 3 imp: 0 hyb: SP2 arom?: 1
9 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
...
13 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
...
Bonds:
...
6 5->7 order: a conj?: 1 aromatic?: 1
7 7->8 order: 1 stereo: CCW bonds: (14 6 8 15) conj?: 1
8 8->9 order: a conj?: 1 aromatic?: 1
...
14 7->1 order: a dir: wedge conj?: 1 aromatic?: 1
15 13->8 order: a conj?: 1 aromatic?: 1
This tells us that bond 7 is atropisomeric and that when looking down the bond (from atom C7 to atom N8), the rotation direction between bonds 14 and 8 (these are the bonds to the lowest numbered atoms on each of the atropisomeric bond) is counterclockwise (CCW).
Formats supporting atropisomers¶
Atropisomers are supported for molecule parsing and writing in Mol, MRV, and CXSmiles formats. For reactions, atropisomers are supported for in rxn, MRV and CXSmiles formats. Atropisomers can be parsed from a CDXML file.
Enhanced Stereochemistry¶
Atropisomers can be part of Enhanced Stereochemistry Groups (Or, And, or Absolute). This is indicated by marking one or both of the atropisomer bond’s atoms as being in the enhanced Stereo Group
Example:
3D Coordinates for Input of Atropisomers¶
If 3D coordinates are available (and 2D coordinates are not), the atropisomer bond is marked only if one of the neighbor bonds is wedged/hashed. The wedge/hash information is ignored except for signaling the presence of the atropisomer. The actual configuration is determined by the 3D coordinates.
Here’s an example. The drawing at the left shows a 3D structure with a wedged bond on one end of the atropisomer bond. The drawing at the right shows a 2D drawing of the same structure. In both cases, the atropisomer bond is highlighted in red. Notice that the bond wedging in the 3D structure is inconsistent with the 3D coordinates; it’s just used to indicate that there is an atropisomer bond, the actual stereochemistry of the bond is determined by the 3D coordinates.
Interpreting atropisomers without coordinates¶
Just as it is possible to interpret atomic and double bond stereochemistry from SMILES without providing atomic coordinates, the RDKit adopts (starting with the 2024.03.2 release) a convention for interpreting bond wedge information in CXSMILES that do not have coordinates provided.
In order to understand how this representation works, a quick digression is necessary to explain the difference in the way bonds are numbered in CXSMILES and the RDKit.
In the RDKit ring closure bonds are added last; they are the highest numbered bonds in the molecule. In CXSMILES, the ring closure bonds are added immediately upon closing the ring.
Here’s an illustration of this for the SMILES CC1=CC=CC(I)=C1N1C(C)=CC=C1Br; the RDKit bond numbering (what we saw above) is shown first and the CXSMILES numbering is shown second.
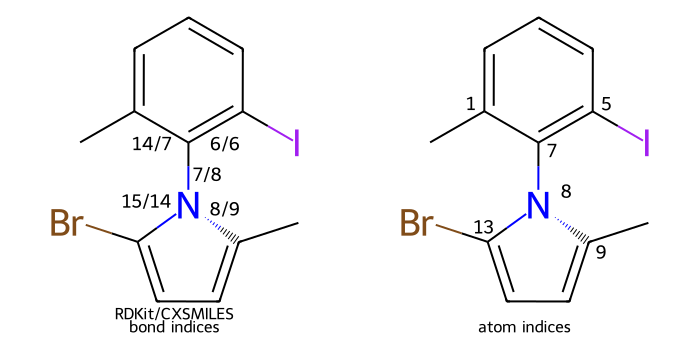
To describe this atropisomer in CXSMILES without using coordinates, we adopt the convention that the atropisomeric bond’s stereochemistry is CCW when the bond to the lowest-numbered neighbor of the start atom
(in this case the neighbor connected via bond 7) is wedged and write the CXSMILES for this molecule as CC1=CC=CC(I)=C1N1C(C)=CC=C1Br |wU:7.7||. The stereo values for all possible combinations are shown here:
from |
to |
bond index |
type |
stereo |
|
|---|---|---|---|---|---|
7 |
1 |
7 |
wedge |
CCW |
|
7 |
5 |
6 |
wedge |
CW |
|
8 |
9 |
9 |
wedge |
CW |
|
8 |
13 |
14 |
wedge |
CCW |
|
7 |
1 |
7 |
dash |
CW |
|
7 |
5 |
6 |
dash |
CCW |
|
8 |
9 |
9 |
dash |
CCW |
|
8 |
13 |
14 |
dash |
CW |
|
It’s also possible to wedge multiple neighbor bonds around an atropisomeric bond, but the wedging must be consistent based on the table above;
so the CXSMILES CC1=CC=CC(I)=C1N1C(C)=CC=C1Br |wD:7.7,wU:8.9| is valid, but CC1=CC=CC(I)=C1N1C(C)=CC=C1Br |wD:7.7,wU:8.14| is not.
Validation of Atropisomers¶
Invalid atropisomers (for example, those with two equivalent groups on one end)
will be removed when reading molecules in with sanitization enabled or by
calling cleanupAtropisomers(mol).
Searching and Canonicalization¶
Atropisomers are supported in the canonicalization algorithm of RDKit. They are ignored in substructure searching and similarity searching at this time.
Query Features in Molecule Drawings¶
Compactly and clearly including information about query features in molecule drawings is a challenging problem. This is definitely a work in progress, but this section describes what is currently supported.
Query Bonds¶
Here is an example image showing how different bond and query-bond types are rendered.
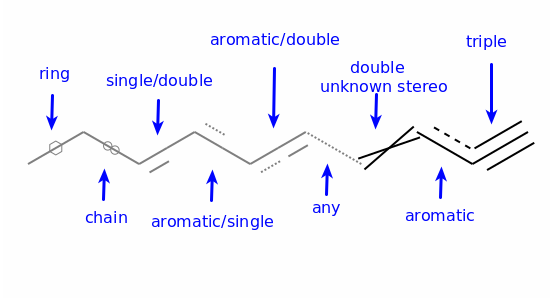
There’s clearly some room for improvement here, for example, it’s not trivial to distinguish “Any” bonds from query bonds where no special handling has been implemented (“other” query types):
Query Atoms¶
At the moment the only real support for atomic query features is rendering of atom lists (and “NOT” atom lists); other atomic queries are rendered with a simple ?:
Conformer Generation¶
Introduction¶
The RDKit can generate conformers for molecules using two different methods. The original method used distance geometry. [16] The default algorithm followed is:
The molecule’s distance bounds matrix is calculated based on the connection table and a set of rules.
The bounds matrix is smoothed using a triangle-bounds smoothing algorithm.
A random distance matrix that satisfies the bounds matrix is generated.
This distance matrix is embedded in 3D dimensions (producing coordinates for each atom).
The resulting coordinates are cleaned up somewhat using the “distance geometry force field”, based on distance constraints from the bounds matrix.
The RDKit also has an implementation of the ETKDG method of Riniker and Landrum [17] which modifies step 5 above to also use torsion angle preferences from the Cambridge Structural Database (CSD) to correct the conformers after distance geometry has been used to generate them. The ETDKDG approach can be extended to include additional torsion terms for small rings and/or macrocycles [18].
When using the ETKDG approaches the quality of the conformers generated is generally good enough to allow them to be used “as is” (i.e. without a subsequent minimization step with another force field) for many applications.
Parameters Controlling Conformer Generation¶
A large number of parameters which allow control over the conformer generation
process are available in the EmbedParameters class. A subset of particularly
useful parameters are described here:
randomSeed: (default -1) allows you to set a random seed to allow reproducible resultsnumThreads: (default 1) sets the number of compute threads to be used when generating multiple conformers. If set to 0 this will use the maximum number of threads allowed on your system.useRandomCoords: (default False) if set to True then random-coordinate embedding will be done: instead of steps 3. and 4. above, the atoms will be randomly placed in a box and then their positions will be minimized with the “distance geometry force field” in step 5. This approach was described in reference [19]enforceChirality: (default True) ensures that the chirality of specified stereocenters in the molecule is preserved in the conformers.embedFragsSeparately: (default True) for molecules made up of multiple disconnected fragments, this cause conformers of the fragments to be generated independently of each other.coordMap: (default empty) can be used to provide 3D coordinates which will be used to constrain the positions of some of the atoms in the molecule.boundsMat: (default empty) can be used to provide the distance bounds matrix for the molecule.useExpTorsionAnglePrefs: (default False) use the ET part of ETKDG [17]useBasicKnowledge: (default False) use the K part of ETKDG [17]ETVersion: (default 1) specify the version of the standard torsion definitions to use. NOTE for both ETKDGv2 and ETKDGv3 this should be 2 since ETKDGv3 uses the ETKDGv2 definitions for standard torsions (apologies for the confusing numbering)useSmallRingTorsions: (default False) use the sr part of srETDKGv3 [18]useMacrocycleTorsions: (default False) use the macrocycle torsions from ETKDGv3 [18]useMacrocycle14config: (default False) use the 1-4 distance bounds from ETKDGv3 [18]forceTransAmides: (default True) constrain amide bonds to be transpruneRMsThresh: (default -1.0) if >0.0 this turns on RMSD pruning of the conformersonlyHeavyAtomsForRMS: (default: False) toggles ignoring H atoms when doing RMSD pruninguseSymmetryForPruning: (default True) uses symmetry to calculate the minimum RMSD between two conformers when doing RMSD pruning. Note that enabling this causes the RMSD computation to act as if onlyHeavyAtomsForRMS is set to true (even if the parameter itself is set to False).
Note that there are pre-configured parameter objects for the available ETKDG
versions: ETKDG, ETKDGv2, ETKDGv3, and srETKDGv3
Additional Information About the Fingerprints¶
This section, which is not currently comprehensive, is intended to provide some documentation about the types of fingerprints available in the RDKit. We don’t reproduce information that can be found in the literature, but try and capture the unpublished bits (of which there are quite a few).
RDKit Fingerprints¶
This is an RDKit-specific fingerprint that is inspired by (though it differs significantly from) public descriptions of the Daylight fingerprint [7]. The fingerprinting algorithm identifies all subgraphs in the molecule within a particular range of sizes, hashes each subgraph to generate a raw bit ID, mods that raw bit ID to fit in the assigned fingerprint size, and then sets the corresponding bit. Options are available to generate count-based forms of the fingerprint or “non-folded” forms (using a sparse representation).
- The default scheme for hashing subgraphs is to hash the individual bonds based on:
the types of the two atoms. Atom types include the atomic number (mod 128), and whether or not the atom is aromatic.
the degrees of the two atoms in the path.
the bond type (or
AROMATICif the bond is marked as aromatic)
Fingerprint-specific options¶
minPathandmaxPathcontrol the size (in bonds) of the subgraphs/paths considered
nBitsPerHash: If this is greater than one, each subgraph will set more than one bit. The additional bits will be generated by seeding a random number generator with the original raw bit ID and generating the appropriate number of random numbers.
useHs: toggles whether or not Hs are included in the subgraphs/paths (assuming that there are Hs in the molecule graph.
tgtDensity: if this is greater than zero, the fingerprint will be repeatedly folded in half until the density of set bits is greater than or equal to this value or the fingerprint only contains minSize bits. Note that this means that the resulting fingerprint will not necessarily be the size you requested.
branchedPaths: if this is true (the default value), the algorithm will use subgraphs (i.e features can be branched. If false, only linear paths will be considered.
useBondOrder: if true (the default) bond types will be considered when hashing subgraphs, otherwise this component of the hash will be ignored.
Pattern Fingerprints¶
These fingerprints were designed to be used in substructure screening. These
are, as far as I know, unique to the RDKit. The algorithm identifies features in
the molecule by doing substructure searches using a small number (12 in the
2019.03 release of the RDKit) of very generic SMARTS patterns - like
[*]~[*]~[*](~[*])~[*] or [R]~1[R]~[R]~[R]~1, and then hashing each
occurrence of a pattern based on the atom and bond types involved. The fact that
particular pattern matched the molecule at all is also stored by hashing the
pattern ID and size. If a particular feature contains either a query atom or a
query bond (e.g. something generated from SMARTS), the only information that is
hashed is the fact that the generic pattern matched.
For the 2019.03 release, the atom types use just the atomic number of the
atom and the bond types use the bond type, or AROMATIC for aromatic bonds).
NOTE: Because it plays an important role in substructure screenout, the internals of this fingerprint (the generic patterns used and/or the details of the hashing algorithm) may change from one release to the next.
Atom-Pair and Topological Torsion Fingerprints¶
These two related fingerprints are implemented based on the original papers: [8] [9]. Atoms are typed based on atomic number, number of pi electrons, and the degree of the atom. Optionally information about atomic chirality can also be integrated into the atom types. Both fingerprint types can be generated in explicit or sparse form and as bit or count vectors. These fingerprint types are different from the others in the RDKit in that bits in the sparse form of the fingerprint can be directly explained (i.e. the “hashing function” used is fully reversible).
These fingerprints were originally “intended” to be used in count-vectors and they seem to work better that way. The default behavior of the explicit bit-vector forms of both fingerprints is to use a “count simulation” procedure where multiple bits are set for a given feature if it occurs more than once. The default behavior is to use 4 fingerprint bits for each feature (so a 2048 bit fingerprint actually stores information about the same number of features as a 512 bit fingerprint that isn’t using count simulation). The bins correspond to counts of 1, 2, 4, and 8. As an example of how this works: if a feature occurs 5 times in a molecule, the bits corresponding to counts 1, 2, and 4 will be set.
Morgan and Feature Morgan Fingerprints¶
These are implemented based on the original paper [10]. The algorithm follows the description in the paper as closely as possible with the exception of the chemical feature definitions used for the “Feature Morgan” fingerprint - the RDKit implementation uses the feature types Donor, Acceptor, Aromatic, Halogen, Basic, and Acidic with definitions adapted from those in the paper [11]. It is possible to provide your own atom types. The fingerprints are available as either explicit or sparse count vectors or explicit bit vectors.
Layered Fingerprints¶
These are another “RDKit original” and were developed with the intention of using them as a substructure fingerprint. Since the pattern fingerprint is far simpler and has proven to be quite effective as a substructure fingerprint, the layered fingerprint hasn’t received much attention. It may still be interesting for something, so we continue to include it.
The idea of the fingerprint is generate features using the same subgraph (or path) enumeration algorithm used in the RDKit fingerprint. After a subgraph has been generated, it is used to set multiple bits based on different atom and bond type definitions.
Self-Contained Structure Representations (SCSR) for Macromolecules¶
The SCSR support added to RDKit follows the description in the BIOVIA document “biovia_ctfileformats_2020.pdf” available from “CTfile Formats - Dassault Systèmes”. That document does not provide any real detail for that format, and contains one example file.
In addition, Biovia Draw supports reading and writing this format. As much as possible, the RDKit support allows the functionality supported by Biovia Draw. One exception is the RDKit treatment of hydrogen bonds in SCSR files/blocks (vide infra).
Representation¶
Main mol¶
An SCSR file contains a mol with a CTAB in v3000 format. That CTAB can contain elemental atoms and macro atoms corresponding to amino acids (AA), RNA, and DNA elements. The RNA and DNA elements are represented by three parts – a phosphate group, a sugar, and a base.
Each atom line in the CTAB can refer to an elemental atom or a macro atom. Macro atom lines have a text description of the macro name and must have a CLASS and an ATTCHORD attributed. They can also have an optional SEQID attribute.
According to the Biovia doc, the CLASS attribute must have a value that is one of: AA, dAA, DNA, RNA, SUGAR, BASE, PHOSPHATE, LINKER, CHEM, LGRP, MODAA, MODdAA, MODDNA, MODRNA, XLINKAA, XLINKdAA, XLINKDNA, XLINKRNA. For an SCSR mol block, (and for any SGROUP), RDKit requires that the CLASS attribute be one of these values.
The SEQID is a sequential integer and is ignored by this treatment. Typically, the three parts of an RNA or DNA element have the same SEQID.
The ATTCHORD attribute must have a specification for each bond that comes from the macro atom. The specification is contained between parentheses, and the first character is a integer that indicates the number of items that follow. Each connection is specified with the atom ID (1 offset) for the connected atom in the main CTAB, and a string that indicates the attachment point for this macro atom. The attachment point string is always two characters. The first indicates the order, and is a capital letter starting at A. The second char is one of “l” (left), “r” (right), “x” (cross connections – e.g. for cysteine). In this implementation, the second character can also be “h” for hydrogen bond. Thus, the attach connections are almost always in the list: “Al”, “Br”, “Cx” or “Ch”. For example:
ATTCHORD=(6 15 Br 13 Al 21 Cx)
Templates¶
Template Header¶
In addition to the CTAB for the main molecule, each macro atom is detailed automatically in a TEMPLATE. The TEMPLATES are numbered and appear between a BEGIN TEMPLATES line and an END TEMPLATES line. Each template starts with a TEMPLATE line that indicates the template number (1 to n), the template Class and Name, and the NATREPLACE attribute.
The Class and Name consists of three (or more) parts separated by forward slashes. The first part is the CLASS, and must match the CLASS attribute in one or more of the atoms in the main CTAB. The second and third (and subsequent) items are choices for names of the macro atom template. Typically, the first name is the long form and second is the short form, as in “AA/Ala/A” for alanine. This treatment does not enforce any restrictions on the name lengths. The name given the macro atom in the main CTAB must match one of the names in one of the templates with the correct class. The NATREPLACE attribute specifies the natural replacement for this macro atom, and is ignored by this treatment. Example:
M V30 TEMPLATE 1 SUGAR/Rib/R NATREPLACE=SUGAR/R
Main Template CTAB and SGROUPs¶
After the TEMPLATE is a full CTAB with the atoms and bonds of the template. Each CTAB must contain an SGROUP for the main macro definition atoms and bonds, and one for each leaving group. The main SGROUP for the template must have a LABEL attribute that is the same as one of the names in the TEMPLATE line, and it must have a CLASS attribute that matches the TEMPLATE line class.
In addition, the main SGROUP must have ATOMS, XBONDS, NATREPLACE attributes. ATOMS is a list of all atom IDs (1-offset). XBONDS are the IDs of the connecting bonds. NATRELACE is the natural replacement as discussed above. The XBONDS and NATREPLACE attributes are ignored by this treatment.
The main SGROUP must also contain a list of SAP (SGROUP attachment points) attributes. Each one is enclosed in parentheses, and starts with a “3”. That is followed by two atom IDs of a bond that starts in this SGROUP and goes to an atom NOT in this SGROUP, and this is followed by a string indicating the connection name. The connection name, as discussed above, is usually one of “Al”, “Br”, “Cx” or “Ch”. SAPs “Al”, “Br”, “Cx” will have one SAP, and “Ch” will typically have two or three distinct SAP attributes. The order of the SAP attributes for hydrogen bonds (“Ch”) matters, and should go from the H-bond atom nearest to the connecting “Al” atom to the most distant. Hydrogen bonds do not have a leaving group, so the second ID of the designation is “0”.
Example:
M V30 2 SUP 2 ATOMS=(8 1 2 3 4 5 6 7 8) XBONDS=(1 7)-
M V30 LABEL=U CLASS=BASE SAP=(3 4 9 Al) SAP=(3 8 0 Ch) SAP=(3 6 0 Ch) SAP=(3 7 0 Ch)-
NATREPLACE=BASE/U
In addition to the main SGROUP, there will be an SGROUP that identifies each leaving group. Most leaving groups have one atom, but they could have multiple atoms. The leaving group SGROUPS have ATOMS, XBONDS, LABEL, and CLASS attributes. The XBONDS attribute is ignored in this treatment. LABEL must be “LGRP” (leaving group).
Example:
M V30 1 SUP 1 ATOMS=(1 11) XBONDS=(1 11) LABEL=H CLASS=LGRP
Parsing SCSR files and text blocks¶
RDKit converts the SCSR data into a fully atomistic representation.
An SGROUP is produced in the resulting RWMol for each template and retained leaving group from the SCSRMol. The name of each SGROUP is derived from the template name, the sequence number if present, and, for leaving groups, the leaving group SAP ID (e.g. “Al”, “Br”, “Cx” or “Ch”).
The following calls parse the SCSR mol or block produces a fully atomistic RDKit mol.:
MolFromSCSRDataStream (stream to mol)
MolFromSCSRBlock (SCSR text block to mol)
MolFromSCSRFile (SCSR file to mol)
These functions all take, in addition to the strean, text block, or file name, a MolFileParserParams and a MolFromSCSRParams parameter. The MolFileParserParams parameter is used to control the parsing of the SCSR data, as is describe elsewhere in this document. The MolFromSCSRParams parameter is used to control the conversion of the SCSR data into a full atomistic representation.
MolFromScsrParams has three properties:
The “ScsrTemplateNames” property controls how the Sgoups of the expanded file are generated, and must be one of: ScsrTemplateNamesAsEntered,
ScsrTemplateNamesUseFirstName, or ScsrTemplateNamesUseLastName. If ScsrTemplateNamesAsEntered is specified, the name as referenced in the main SCSR CTAB will be used.
The ”includeLeavingGroups” property controls whether leaving groups that are not replaced in the main CTAB are included in the resulting atomistic file.
The leaving groups that are so retained become end caps and caps on unused cross-link sites. Setting this property to “false” causes the end caps and cross-link caps to remain unsubstituted. This allows the results to be used as a full atom query for the sub-units from the SCSR mol.
The “SCSRBaseHbondOptions” property controls the treatment of hydrogen bonds in the SCSR file. This is described in the following section.
Hydrogen Bonds¶
For sense-antisense pairings in DNA and RNA, the hydrogen bonds are represented as a single hydrogen bond in the SCSR representation. When converted to a full atomistic representation, each such hydrogen bond can represent up to 3 hydrogen bonds between the full-atomistic representations of the Base groups.
The processing of H-bonds is contorlled by the ScsrBaseHbondOptions member of the ScsrMolFileParserParams parameter. The options are:
ScsrBaseHbondOptionsIgnore - if this is selected, all H-bonds are ignored and not processed.
ScsrBaseHbondOptionsUseSapAll = 1 If this is selected, all SAPs for the hbond are used. They must be defined in the template in the correct order, which starts with the first atom nearest the “Al” connection, and continues sequentially
If there are 3 sites on each side and they are complimentary (the donors match acceptors and vice versa), we add the bonds. and we are done. THis is the standard Watson-Crick binding (“https://water.lsbu.ac.uk/water/nucleic_acid_hydration.html”)
If not, we check to check to see if they comply to a wobble bond configuration. There are four generally accepted wobble bonds, and we deal with these four types only. The four known wobble bonds are:
I-C
I-U
I-A
G-U
“I” stands for inosine - it has only two available hbond sites . The pair G-U has three sites on each end, but they are not complimentary. (https://en.wikipedia.org/wiki/Wobble_base_pair#:~:text=A%20wobble%20base%20pair%20is,hypoxanthine%2Dcytosine%20(I%2DC).
For pairs that have I (inosine) or something like it, the configuration must be DA, so those two atoms form the two H bonds. The other side (C,U or A) has confiuration of AAD (C), ADA (U), or DAD (A). For C-type bases and A type bases, the second and third atoms are used (AD), and for U types, the first two atoms are used (AD).
For the GU pair, both sides have 3 atoms, but they are not complimentary. The second and third sites on the G side are used (DA), and the first two sites on the U side are used (AD).
In any other case, we punt and add just one bond between the first hydrogen-bond atom on both sides even if they are NOT complemenary. This just indicates that the sides are h-bonded somehow and keeps the overall pairing straight.
ScsrBaseHbondOptionsUseSapOne If this is selected, only one SAP hbond per base is used. If multiple SAPs are defined, the first hbond site is used even if it is not the best. No attempt is made to match the Donor/Acceptor status of the chosen bond sites. (this just maintains the relationship between the to base pairs)
ScsrBaseHbondOptionsAuto For bases that are C,G,A,T,U,In (and derivatives) the standard Watson-Crick Hbonding is used, and is determined by substructure matching. No Hbond SAPs (“Ch”) need to be defined in the template, and if defined, they are ignored.
Processing of the H-bond sites so determined is done just as it is when ScsrBaseHbondOptionsUseSapAll is selected. (see above).
Example of Peptide conversion:
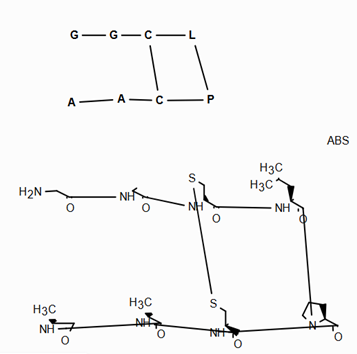
Example of DNA-RNA paired strands conversion:
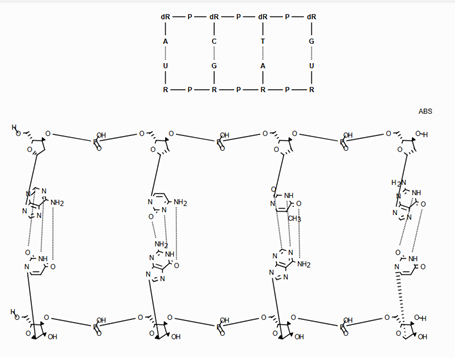
Example of “Wobble” pairing conversion:
Here are two examples of SCSR files:
Feature Flags: global variables affecting RDKit behavior¶
The RDKit uses a number of “feature flags”: global variables which affect its behavior. These have generally been added to maintain backwards compatibility when introducing new algorithms which yield different results.
Here’s are the current feature flags:
preferCoordGen: when this istrueSchrodinger’s open-source Coordgen library will be used to generate 2D coordinates of molecules. The default value isfalse. This can be set from C++ using the variableRDKit::RDDepict::preferCoordGenor from Python using the functionrdDepictor.SetPreferCoordGen(). Added in the 2018.03 release.
allowNontetrahedralChirality: when this istruenon-tetrahedral chirality will be perceived from 3D coordinates. The default value istrueunless the environment variableRDK_ENABLE_NONTETRAHEDRAL_STEREOis set to"0". Can set/checked from C++ using the functionsRDKit::Chirality::setAllowNontetrahedralChirality()/RDKit::Chirality::getAllowNontetrahedralChirality()or from Python using the functionsChem.SetAllowNontetrahedralChirality()/Chem.GetAllowNontetrahedralChirality(). Added in the 2022.09 release.
useLegacyStereoPerception: when this istruethe legacy implementation for perceiving stereochemistry will be used. The default value istrueunless the environment variableRDK_USE_LEGACY_STEREO_PERCEPTIONis set to"0". Can set/checked from C++ using the functionsRDKit::Chirality::setUseLegacyStereoPerception()/RDKit::Chirality::getUseLegacyStereoPerception()or from Python using the functionsChem.SetUseLegacyStereoPerception()/Chem.GetUseLegacyStereoPerception(). Added in the 2022.09 release.
Footnotes
License¶

This document is copyright (C) 2007-2021 by Greg Landrum
This work is licensed under the Creative Commons Attribution-ShareAlike 4.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-sa/4.0/ or send a letter to Creative Commons, 543 Howard Street, 5th Floor, San Francisco, California, 94105, USA.
The intent of this license is similar to that of the RDKit itself. In simple words: “Do whatever you want with it, but please give us some credit.”