RDKit Cookbook¶
Introduction¶
What is this?¶
This document provides example recipes of how to carry out particular tasks using the RDKit functionality from Python. The contents have been contributed by the RDKit community, tested with the latest RDKit release, and then compiled into this document. The RDKit Cookbook is written in reStructuredText, which supports Sphinx doctests, allowing for easier validation and maintenance of the RDKit Cookbook code examples, where appropriate.
What gets included?¶
The examples included come from various online sources such as blogs, shared gists, and the RDKit mailing lists. Generally, only minimal editing is added to the example code/notes for formatting consistency and to incorporate the doctests. We have made a conscious effort to appropriately credit the original source and authors. One of the first priorities of this document is to compile useful short examples shared on the RDKit mailing lists, as these can be difficult to discover. It will take some time, but we hope to expand this document into 100s of examples. As the document grows, it may make sense to prioritize examples included in the RDKit Cookbook based on community demand.
Feedback and Contributing¶
If you have suggestions for how to improve the Cookbook and/or examples you would like included, please contribute directly in the source document (the .rst file). Alternatively, you can also send Cookbook revisions and addition requests to the mailing list: <rdkit-discuss@lists.sourceforge.net> (you will need to subscribe first).
Note
The Index ID# (e.g., RDKitCB_##) is simply a way to track Cookbook entries and image file names. New Cookbook additions are sequentially index numbered, regardless of where they are placed within the document. As such, for reference, the next Cookbook entry is RDKitCB_41.
Drawing Molecules (Jupyter)¶
Include an Atom Index¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
IPythonConsole.ipython_useSVG=True #< set this to False if you want PNGs instead of SVGs
def mol_with_atom_index(mol):
for atom in mol.GetAtoms():
atom.SetAtomMapNum(atom.GetIdx())
return mol
# Test in a kinase inhibitor
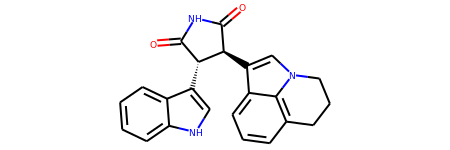
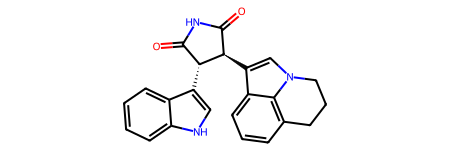
mol = Chem.MolFromSmiles("C1CC2=C3C(=CC=C2)C(=CN3C1)[C@H]4[C@@H](C(=O)NC4=O)C5=CNC6=CC=CC=C65")
# Default
mol
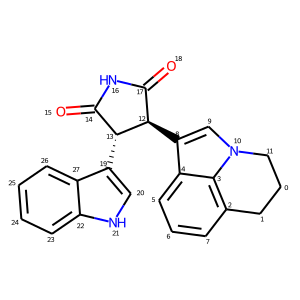
# With atom index
mol_with_atom_index(mol)
In contrast to the approach below, the atom index zero is not displayed.
A simpler way to add atom indices is to adjust the IPythonConsole properties. This produces a similar image to the example above, the difference being that the atom indices are now near the atom, rather than at the atom position.
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
IPythonConsole.drawOptions.addAtomIndices = True
IPythonConsole.molSize = 300,300
mol = Chem.MolFromSmiles("C1CC2=C3C(=CC=C2)C(=CN3C1)[C@H]4[C@@H](C(=O)NC4=O)C5=CNC6=CC=CC=C65")
mol
Include a Bond Index¶
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.ipython_useSVG=True #< set this to False if you want PNGs instead of SVGs
# Test in a kinase inhibitor
mol = Chem.MolFromSmiles("C1CC2=C3C(=CC=C2)C(=CN3C1)[C@H]4[C@@H](C(=O)NC4=O)C5=CNC6=CC=CC=C65")
# Default
mol
# Add bond indices
IPythonConsole.drawOptions.addBondIndices = True
IPythonConsole.molSize = 350,300
mol
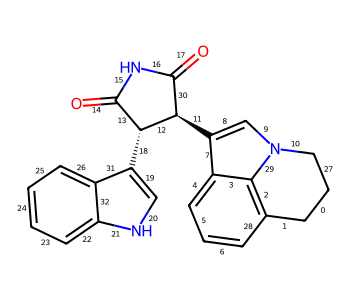
Include a Calculation¶
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.molSize = 250,250
m = Chem.MolFromSmiles('c1ncncc1C(=O)[O-]')
AllChem.ComputeGasteigerCharges(m)
m
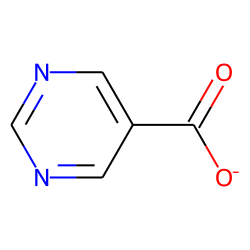
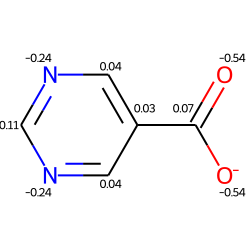
m2 = Chem.Mol(m)
for at in m2.GetAtoms():
lbl = '%.2f'%(at.GetDoubleProp("_GasteigerCharge"))
at.SetProp('atomNote',lbl)
m2
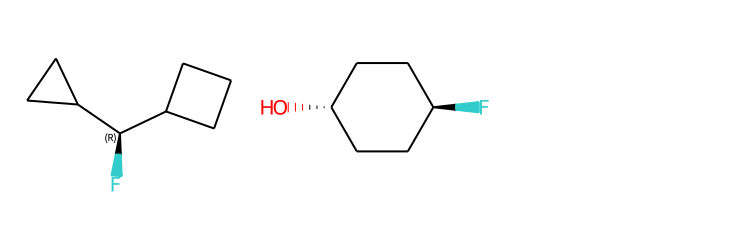
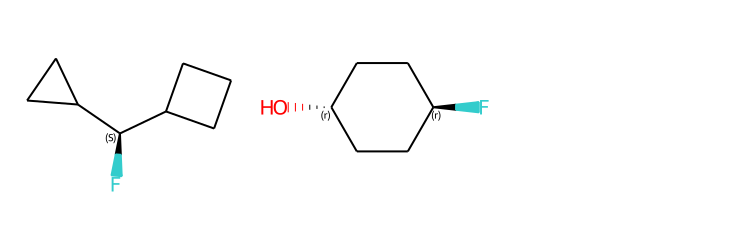
Include Stereo Annotations¶
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.drawOptions.addAtomIndices = False
IPythonConsole.drawOptions.addStereoAnnotation = True
# Default Representation uses legacy FindMolChiralCenters() code
m1 = Chem.MolFromSmiles('C1CC1[C@H](F)C1CCC1')
m2 = Chem.MolFromSmiles('F[C@H]1CC[C@H](O)CC1')
Draw.MolsToGridImage((m1,m2), subImgSize=(250,250))
# new stereochemistry code with more accurate CIP labels, 2020.09 release
from rdkit.Chem import rdCIPLabeler
rdCIPLabeler.AssignCIPLabels(m1)
rdCIPLabeler.AssignCIPLabels(m2)
Draw.MolsToGridImage((m1,m2), subImgSize=(250,250))
Black and White Molecules¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
ms = [Chem.MolFromSmiles(x) for x in ('Cc1onc(-c2ccccc2)c1C(=O)N[C@@H]1C(=O)N2[C@@H](C(=O)O)C(C)(C)S[C@H]12','CC1(C)SC2C(NC(=O)Cc3ccccc3)C(=O)N2C1C(=O)O.[Na]')]
Draw.MolsToGridImage(ms)
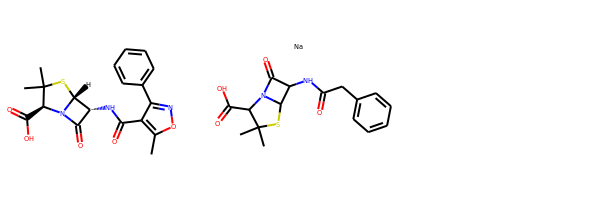
IPythonConsole.drawOptions.useBWAtomPalette()
Draw.MolsToGridImage(ms)
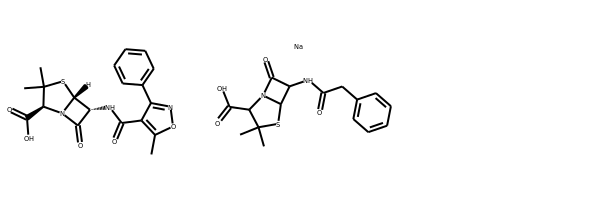
# Alternatively, use the rdMolDraw2D package
from rdkit.Chem.Draw import rdMolDraw2D
import io
from PIL import Image
drawer = rdMolDraw2D.MolDraw2DCairo(500,180,200,180)
drawer.drawOptions().useBWAtomPalette()
drawer.DrawMolecules(ms)
drawer.FinishDrawing()
bio = io.BytesIO(drawer.GetDrawingText())
Image.open(bio)
# works for reactions too:
# rxn is from https://github.com/rdkit/UGM_2020/blob/master/Notebooks/Landrum_WhatsNew.ipynb
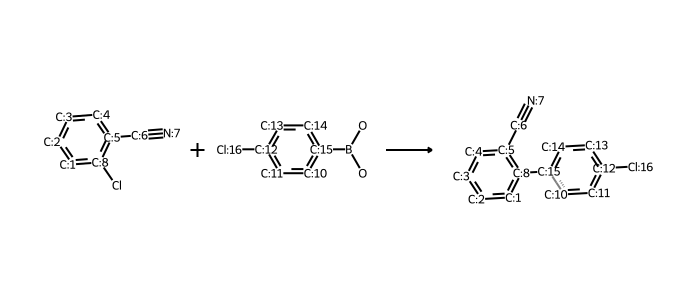
from rdkit.Chem import rdChemReactions
rxn = rdChemReactions.ReactionFromSmarts("[cH:1]:1:[cH:2]:[cH:3]:[cH:4]:[cH:5](-[C:6]#[N:7]):[c:8]:1-[Cl].\
[cH:10]:1:[cH:11]:[cH:12](-[Cl:16]):[cH:13]:[cH:14]:[cH:15]:1-B(-O)-O>>\
[cH:1]:1:[cH:2]:[cH:3]:[cH:4]:[cH:5](-[C:6]#[N:7]):[c:8]:1-[cH:15]:1[cH:10]:[cH:11]:[cH:12](-[Cl:16]):[cH:13]:[cH:14]:1")
drawer = rdMolDraw2D.MolDraw2DCairo(700,300)
drawer.drawOptions().useBWAtomPalette()
drawer.DrawReaction(rxn)
drawer.FinishDrawing()
bio = io.BytesIO(drawer.GetDrawingText())
Image.open(bio)
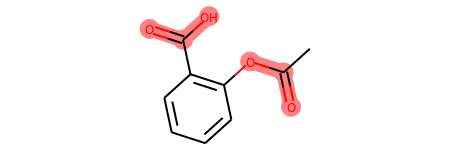
Highlight a Substructure in a Molecule¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
m = Chem.MolFromSmiles('c1cc(C(=O)O)c(OC(=O)C)cc1')
substructure = Chem.MolFromSmarts('C(=O)O')
print(m.GetSubstructMatches(substructure))
((3, 4, 5), (8, 9, 7))
m
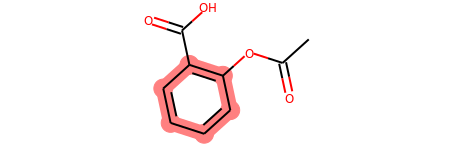
# you can also manually set the atoms that should be highlighted:
m.__sssAtoms = [0,1,2,6,11,12]
m
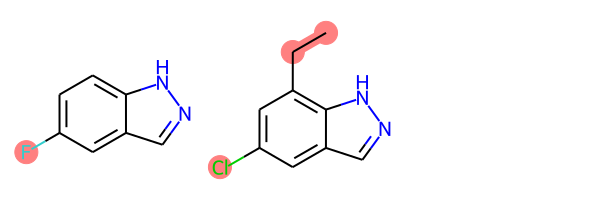
Highlight Molecule Differences¶
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import rdFMCS
from rdkit.Chem.Draw import rdDepictor
rdDepictor.SetPreferCoordGen(True)
IPythonConsole.drawOptions.minFontSize=20
mol1 = Chem.MolFromSmiles('FC1=CC=C2C(=C1)C=NN2')
mol2 = Chem.MolFromSmiles('CCC1=C2NN=CC2=CC(Cl)=C1')
Draw.MolsToGridImage([mol1, mol2])
def view_difference(mol1, mol2):
mcs = rdFMCS.FindMCS([mol1,mol2])
mcs_mol = Chem.MolFromSmarts(mcs.smartsString)
match1 = mol1.GetSubstructMatch(mcs_mol)
target_atm1 = []
for atom in mol1.GetAtoms():
if atom.GetIdx() not in match1:
target_atm1.append(atom.GetIdx())
match2 = mol2.GetSubstructMatch(mcs_mol)
target_atm2 = []
for atom in mol2.GetAtoms():
if atom.GetIdx() not in match2:
target_atm2.append(atom.GetIdx())
return Draw.MolsToGridImage([mol1, mol2],highlightAtomLists=[target_atm1, target_atm2])
view_difference(mol1,mol2)
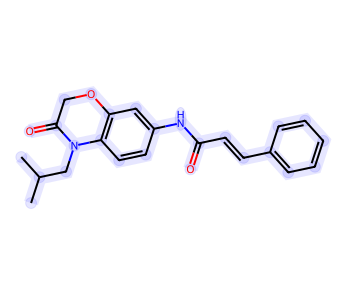
Highlight Entire Molecule¶
from rdkit import Chem
from rdkit.Chem.Draw import rdMolDraw2D
import io
from PIL import Image
mol = Chem.MolFromSmiles('CC(C)CN1C(=O)COC2=C1C=CC(=C2)NC(=O)/C=C/C3=CC=CC=C3')
rgba_color = (0.0, 0.0, 1.0, 0.1) # transparent blue
atoms = []
for a in mol.GetAtoms():
atoms.append(a.GetIdx())
bonds = []
for bond in mol.GetBonds():
aid1 = atoms[bond.GetBeginAtomIdx()]
aid2 = atoms[bond.GetEndAtomIdx()]
bonds.append(mol.GetBondBetweenAtoms(aid1,aid2).GetIdx())
drawer = rdMolDraw2D.MolDraw2DCairo(350,300)
drawer.drawOptions().fillHighlights=True
drawer.drawOptions().setHighlightColour((rgba_color))
drawer.drawOptions().highlightBondWidthMultiplier=20
drawer.drawOptions().clearBackground = False
rdMolDraw2D.PrepareAndDrawMolecule(drawer, mol, highlightAtoms=atoms, highlightBonds=bonds)
bio = io.BytesIO(drawer.GetDrawingText())
Image.open(bio)
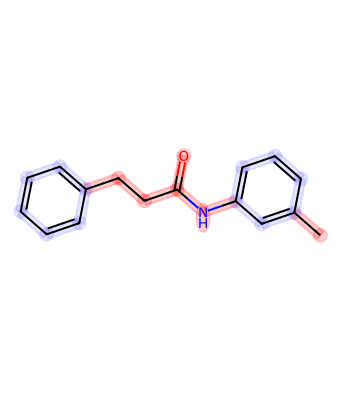
Highlight Molecule with Multiple Colors¶
from rdkit import Chem
from rdkit.Chem.Draw import rdMolDraw2D
import io
from PIL import Image
from collections import defaultdict
mol = Chem.MolFromSmiles('CC1=CC(=CC=C1)NC(=O)CCC2=CC=CC=C2')
colors = [(0.0, 0.0, 1.0, 0.1), (1.0, 0.0, 0.0, 0.2)]
athighlights = defaultdict(list)
arads = {}
for a in mol.GetAtoms():
if a.GetIsAromatic():
aid = a.GetIdx()
athighlights[aid].append(colors[0])
arads[aid] = 0.3
else:
aid = a.GetIdx()
athighlights[aid].append(colors[1])
arads[aid] = 0.3
bndhighlights = defaultdict(list)
for bond in mol.GetBonds():
aid1 = bond.GetBeginAtomIdx()
aid2 = bond.GetEndAtomIdx()
if bond.GetIsAromatic():
bid = mol.GetBondBetweenAtoms(aid1,aid2).GetIdx()
bndhighlights[bid].append(colors[0])
else:
bid = mol.GetBondBetweenAtoms(aid1,aid2).GetIdx()
bndhighlights[bid].append(colors[1])
d2d = rdMolDraw2D.MolDraw2DCairo(350,400)
d2d.DrawMoleculeWithHighlights(mol,"",dict(athighlights),dict(bndhighlights),arads,{})
d2d.FinishDrawing()
bio = io.BytesIO(d2d.GetDrawingText())
Image.open(bio)
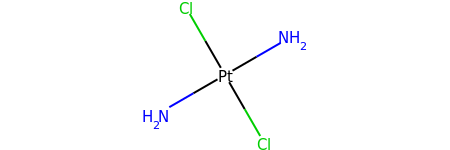
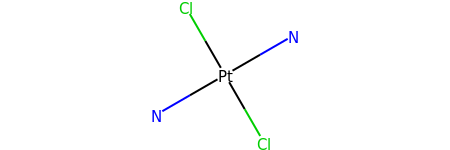
Without Implicit Hydrogens¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
m = Chem.MolFromSmiles('[Pt](Cl)(Cl)(N)N')
m
for atom in m.GetAtoms():
atom.SetProp("atomLabel", atom.GetSymbol())
m
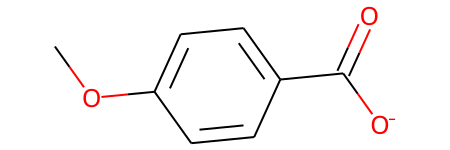
With Abbreviations¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
from rdkit.Chem import rdAbbreviations
m = Chem.MolFromSmiles('COc1ccc(C(=O)[O-])cc1')
m
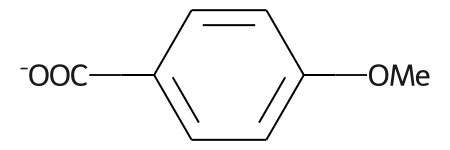
abbrevs = rdAbbreviations.GetDefaultAbbreviations()
nm = rdAbbreviations.CondenseMolAbbreviations(m,abbrevs)
nm
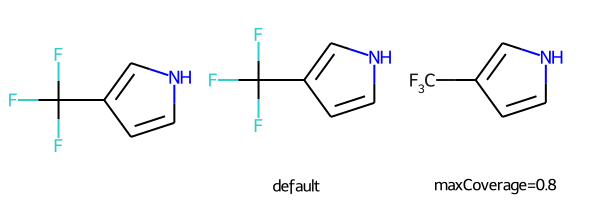
# abbreviations that cover more than 40% of the molecule won't be applied by default
m = Chem.MolFromSmiles('c1c[nH]cc1C(F)(F)F')
nm1 = rdAbbreviations.CondenseMolAbbreviations(m,abbrevs)
nm2 = rdAbbreviations.CondenseMolAbbreviations(m,abbrevs,maxCoverage=0.8)
Draw.MolsToGridImage((m,nm1,nm2),legends=('','default','maxCoverage=0.8'))
# See available abbreviations and their SMILES
# where * is the dummy atom that the group would attach to
abbrevs = rdAbbreviations.GetDefaultAbbreviations()
labels = ["Abbrev", "SMILES"]
line = '--------'
print(f"{labels[0]:<10} {labels[1]}")
print(f"{line:<10} {line}")
for a in abbrevs:
print(f"{a.label:<10} {Chem.MolToSmiles(a.mol)}")
Abbrev SMILES
-------- --------
CO2Et *C(=O)OCC
COOEt *C(=O)OCC
OiBu *OCC(C)C
nDec *CCCCCCCCCC
nNon *CCCCCCCCC
nOct *CCCCCCCC
nHept *CCCCCCC
nHex *CCCCCC
nPent *CCCCC
iPent *C(C)CCC
tBu *C(C)(C)C
iBu *C(C)CC
nBu *CCCC
iPr *C(C)C
nPr *CCC
Et *CC
NCF3 *NC(F)(F)F
CF3 *C(F)(F)F
CCl3 *C(Cl)(Cl)Cl
CN *C#N
NC *[N+]#[C-]
N(OH)CH3 *N(C)[OH]
NO2 *[N+](=O)[O-]
NO *N=O
SO3H *S(=O)(=O)[OH]
CO2H *C(=O)[OH]
COOH *C(=O)[OH]
OEt *OCC
OAc *OC(C)=O
NHAc *NC(C)=O
Ac *C(C)=O
CHO *C=O
NMe *NC
SMe *SC
OMe *OC
CO2- *C(=O)[O-]
COO- *C(=O)[O-]
Using CoordGen Library¶
Some molecules like macrocycles are not represented well using the default RDKit drawing code. As a result, it may be preferable to use the CoordGen integration.
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.molSize = 350,300
from rdkit.Chem import Draw
# default drawing
mol = Chem.MolFromSmiles("C/C=C/CC(C)C(O)C1C(=O)NC(CC)C(=O)N(C)CC(=O)N(C)C(CC(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(CC(C)C)C(=O)NC(C)C(=O)NC(C)C(=O)N(C)C(CC(C)C)C(=O)N(C)C(CC(C)C)C(=O)N(C)C(C(C)C)C(=O)N1C")
mol
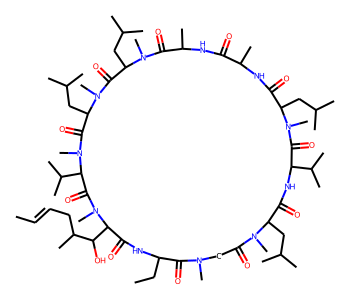
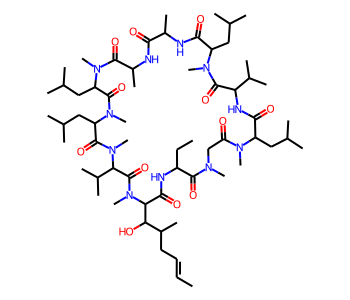
# with CoordGen
from rdkit.Chem import rdCoordGen
rdCoordGen.AddCoords(mol)
mol
It is also possible to use CoordGen with the MolDraw2D class. Here is one way to do that:
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import rdMolDraw2D
from rdkit.Chem import rdDepictor
rdDepictor.SetPreferCoordGen(True)
from rdkit.Chem.Draw import IPythonConsole
from IPython.display import SVG
mol = Chem.MolFromSmiles("C/C=C/CC(C)C(O)C1C(=O)NC(CC)C(=O)N(C)CC(=O)N(C)C(CC(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(CC(C)C)C(=O)NC(C)C(=O)NC(C)C(=O)N(C)C(CC(C)C)C(=O)N(C)C(CC(C)C)C(=O)N(C)C(C(C)C)C(=O)N1C")
drawer = rdMolDraw2D.MolDraw2DSVG(300,300)
drawer.drawOptions().addStereoAnnotation = False
drawer.DrawMolecule(mol)
drawer.FinishDrawing()
SVG(drawer.GetDrawingText())

On a Plot¶
import matplotlib.pyplot as plt
import numpy as np
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
x = np.arange(0, 180, 1)
y = np.sin(x)
mol = Chem.MolFromSmiles('C1CNCCC1C(=O)C')
im = Chem.Draw.MolToImage(mol)
fig = plt.figure(figsize=(10,5))
plt.plot(x, y)
plt.ylim(-1, 5)
ax = plt.axes([0.6, 0.47, 0.38, 0.38], frameon=True)
ax.imshow(im)
ax.axis('off')
# plt.show() # commented out to avoid creating plot with doctest
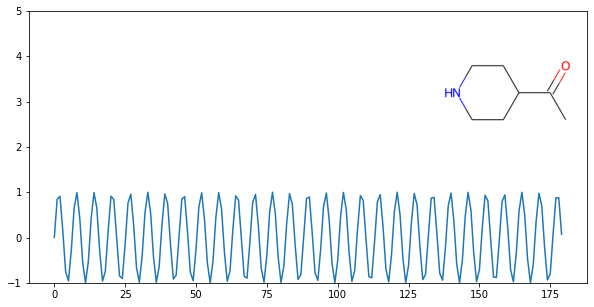
Bonds and Bonding¶
Hybridization Type and Count¶
from rdkit import Chem
m = Chem.MolFromSmiles("CN1C=NC2=C1C(=O)N(C(=O)N2C)C")
for x in m.GetAtoms():
print(x.GetIdx(), x.GetHybridization())
0 SP3
1 SP2
2 SP2
3 SP2
4 SP2
5 SP2
6 SP2
7 SP2
8 SP2
9 SP2
10 SP2
11 SP2
12 SP3
13 SP3
# if you want to count hybridization type (e.g., SP3):
from rdkit import Chem
m = Chem.MolFromSmiles("CN1C=NC2=C1C(=O)N(C(=O)N2C)C")
print(sum((x.GetHybridization() == Chem.HybridizationType.SP3) for x in m.GetAtoms()))
3
Rings, Aromaticity, and Kekulization¶
Count Ring Systems¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
def GetRingSystems(mol, includeSpiro=False):
ri = mol.GetRingInfo()
systems = []
for ring in ri.AtomRings():
ringAts = set(ring)
nSystems = []
for system in systems:
nInCommon = len(ringAts.intersection(system))
if nInCommon and (includeSpiro or nInCommon>1):
ringAts = ringAts.union(system)
else:
nSystems.append(system)
nSystems.append(ringAts)
systems = nSystems
return systems
mol = Chem.MolFromSmiles('CN1C(=O)CN=C(C2=C1C=CC(=C2)Cl)C3=CC=CC=C3')
print(GetRingSystems(mol))
[{1, 2, 4, 5, 6, 7, 8, 9, 10, 11, 12}, {14, 15, 16, 17, 18, 19}]
# Draw molecule with atom index (see RDKitCB_0)
def mol_with_atom_index(mol):
for atom in mol.GetAtoms():
atom.SetAtomMapNum(atom.GetIdx())
return mol
mol_with_atom_index(mol)
Identify Aromatic Rings¶
from rdkit import Chem
m = Chem.MolFromSmiles('c1cccc2c1CCCC2')
m
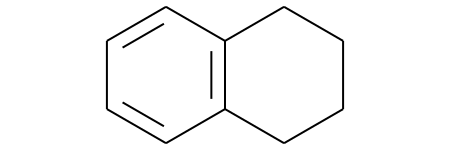
ri = m.GetRingInfo()
# You can interrogate the RingInfo object to tell you the atoms that make up each ring:
print(ri.AtomRings())
((0, 5, 4, 3, 2, 1), (6, 7, 8, 9, 4, 5))
# or the bonds that make up each ring:
print(ri.BondRings())
((9, 4, 3, 2, 1, 0), (6, 7, 8, 10, 4, 5))
# To detect aromatic rings, I would loop over the bonds in each ring and
# flag the ring as aromatic if all bonds are aromatic:
def isRingAromatic(mol, bondRing):
for id in bondRing:
if not mol.GetBondWithIdx(id).GetIsAromatic():
return False
return True
print(isRingAromatic(m, ri.BondRings()[0]))
True
print(isRingAromatic(m, ri.BondRings()[1]))
False
Identify Aromatic Atoms¶
from rdkit import Chem
mol = Chem.MolFromSmiles("c1ccccc1C=CCC")
aromatic_carbon = Chem.MolFromSmarts("c")
print(mol.GetSubstructMatches(aromatic_carbon))
((0,), (1,), (2,), (3,), (4,), (5,))
# The RDKit includes a SMARTS extension that allows hybridization queries,
# here we query for SP2 aliphatic carbons:
olefinic_carbon = Chem.MolFromSmarts("[C^2]")
print(mol.GetSubstructMatches(olefinic_carbon))
((6,), (7,))
There is also an alternative, more efficient approach, using the rdqueries module:
from rdkit import Chem
from rdkit.Chem import rdqueries
mol = Chem.MolFromSmiles("c1ccccc1C=CCC")
q = rdqueries.IsAromaticQueryAtom()
print([x.GetIdx() for x in mol.GetAtomsMatchingQuery(q)])
[0, 1, 2, 3, 4, 5]
q = rdqueries.HybridizationEqualsQueryAtom(Chem.HybridizationType.SP2)
print([x.GetIdx() for x in mol.GetAtomsMatchingQuery(q)])
[0, 1, 2, 3, 4, 5, 6, 7]
qcombined = rdqueries.IsAliphaticQueryAtom()
qcombined.ExpandQuery(q)
print([x.GetIdx() for x in mol.GetAtomsMatchingQuery(qcombined)])
[6, 7]
Stereochemistry¶
Identifying Stereochemistry¶
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.drawOptions.addAtomIndices = True
IPythonConsole.drawOptions.addStereoAnnotation = False
IPythonConsole.molSize = 200,200
m = Chem.MolFromSmiles("C[C@H]1CCC[C@@H](C)[C@@H]1Cl")
m
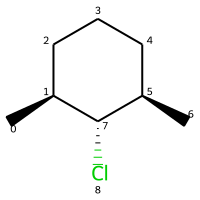
# legacy FindMolChiralCenters()
print(Chem.FindMolChiralCenters(m,force=True,includeUnassigned=True,useLegacyImplementation=True))
[(1, 'S'), (5, 'R'), (7, 'R')]
# new stereochemistry code
print(Chem.FindMolChiralCenters(m,force=True,includeUnassigned=True,useLegacyImplementation=False))
[(1, 'S'), (5, 'R'), (7, 'r')]
# Identifying Double Bond Stereochemistry
IPythonConsole.molSize = 250,250
mol = Chem.MolFromSmiles(r"C\C=C(/F)\C(=C\F)\C=C")
mol
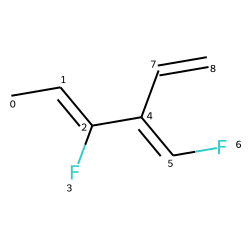
# Using GetStereo()
for b in mol.GetBonds():
print(b.GetBeginAtomIdx(),b.GetEndAtomIdx(),
b.GetBondType(),b.GetStereo())
0 1 SINGLE STEREONONE
1 2 DOUBLE STEREOZ
2 3 SINGLE STEREONONE
2 4 SINGLE STEREONONE
4 5 DOUBLE STEREOE
5 6 SINGLE STEREONONE
4 7 SINGLE STEREONONE
7 8 DOUBLE STEREONONE
# Double bond configuration can also be identified with new
# stereochemistry code using Chem.FindPotentialStereo()
si = Chem.FindPotentialStereo(mol)
for element in si:
print(f' Type: {element.type}, Which: {element.centeredOn}, Specified: {element.specified}, Descriptor: {element.descriptor} ')
Type: Bond_Double, Which: 1, Specified: Specified, Descriptor: Bond_Cis
Type: Bond_Double, Which: 4, Specified: Specified, Descriptor: Bond_Trans
Manipulating Molecules¶
Create Fragments¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole, MolsToGridImage
# I have put explicit bonds in the SMILES definition to facilitate comprehension:
mol = Chem.MolFromSmiles("O-C-C-C-C-N")
mol1 = Chem.Mol(mol)
mol2 = Chem.Mol(mol)
mol1
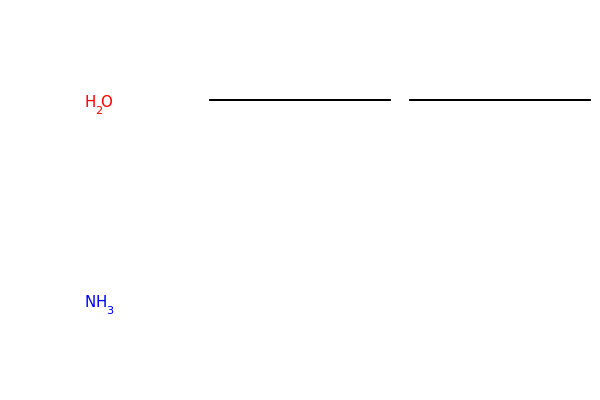
# Chem.FragmentOnBonds() will fragment all specified bond indices at once, and return a single molecule
# with all specified cuts applied. By default, addDummies=True, so empty valences are filled with dummy atoms:
mol1_f = Chem.FragmentOnBonds(mol1, (0, 2, 4))
mol1_f
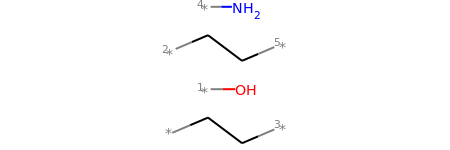
# This molecule can be split into individual fragments using Chem.GetMolFrags():
MolsToGridImage(Chem.GetMolFrags(mol1_f, asMols=True))
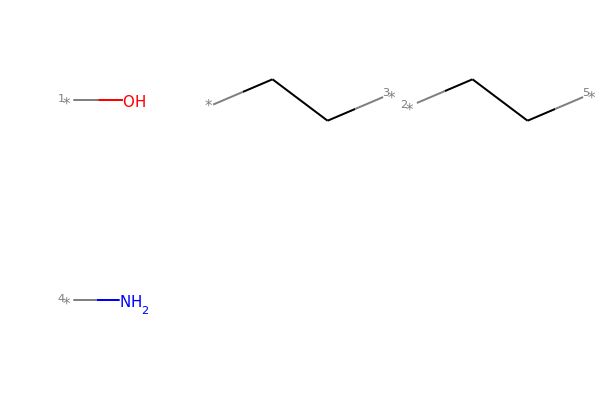
# Chem.FragmentOnSomeBonds() will fragment according to all permutations of numToBreak bonds at a time
# (numToBreak defaults to 1), and return tuple of molecules with numToBreak cuts applied. By default,
# addDummies=True, so empty valences are filled with dummy atoms:
mol2_f_tuple = Chem.FragmentOnSomeBonds(mol2, (0, 2, 4))
mol2_f_tuple[0]
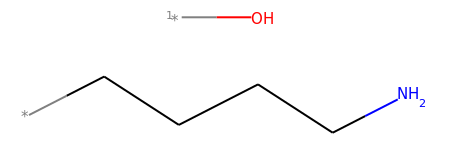
mol2_f_tuple[1]
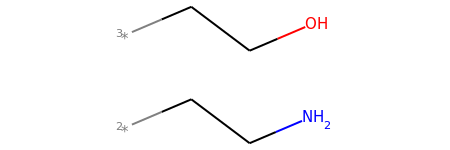
mol2_f_tuple[2]
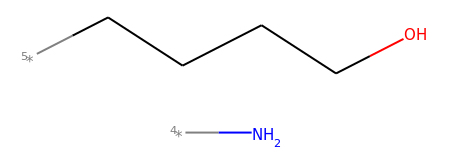
# Finally, you can manually cut bonds using Chem.RWMol.RemoveBonds:
with Chem.RWMol(mol) as rwmol:
for b_idx in [0, 2, 4]:
b = rwmol.GetBondWithIdx(b_idx)
rwmol.RemoveBond(b.GetBeginAtomIdx(), b.GetEndAtomIdx())
# And then call Chem.GetMolFrags() to get sanitized fragments where empty valences were filled with implicit hydrogens:
MolsToGridImage(Chem.GetMolFrags(rwmol, asMols=True))
Largest Fragment¶
from rdkit import Chem
from rdkit.Chem import rdmolops
mol = Chem.MolFromSmiles('CCOC(=O)C(C)(C)OC1=CC=C(C=C1)Cl.CO.C1=CC(=CC=C1C(=O)N[C@@H](CCC(=O)O)C(=O)O)NCC2=CN=C3C(=N2)C(=O)NC(=N3)N')
mol_frags = rdmolops.GetMolFrags(mol, asMols = True)
largest_mol = max(mol_frags, default=mol, key=lambda m: m.GetNumAtoms())
print(Chem.MolToSmiles(largest_mol))
Nc1nc2ncc(CNc3ccc(C(=O)N[C@@H](CCC(=O)O)C(=O)O)cc3)nc2c(=O)[nH]1
The same result can also be achieved with MolStandardize:
from rdkit import Chem
from rdkit.Chem.MolStandardize import rdMolStandardize
mol = Chem.MolFromSmiles('CCOC(=O)C(C)(C)OC1=CC=C(C=C1)Cl.CO.C1=CC(=CC=C1C(=O)N[C@@H](CCC(=O)O)C(=O)O)NCC2=CN=C3C(=N2)C(=O)NC(=N3)N')
# setup standardization module
largest_Fragment = rdMolStandardize.LargestFragmentChooser()
largest_mol = largest_Fragment.choose(mol)
print(Chem.MolToSmiles(largest_mol))
Nc1nc2ncc(CNc3ccc(C(=O)N[C@@H](CCC(=O)O)C(=O)O)cc3)nc2c(=O)[nH]1
Sidechain-Core Enumeration¶
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem import AllChem
# core is '*c1c(C)cccc1(O)'
# chain is 'CN*'
rxn = AllChem.ReactionFromSmarts('[c:1][#0].[#0][*:2]>>[c:1]-[*:2]')
reacts = (Chem.MolFromSmiles('*c1c(C)cccc1(O)'),Chem.MolFromSmiles('CN*'))
products = rxn.RunReactants(reacts) # tuple
print(len(products))
1
print(len(products[0]))
1
print(Chem.MolToSmiles(products[0][0])) # [0][0] to index out the rdchem mol object
CNc1c(C)cccc1O
# The above reaction-based approach is flexible, however if you can generate your
# sidechains in such a way that the atom you want to attach to the core
# is the first one (atom zero), there's a somewhat easier way to do this
# kind of simple replacement:
core = Chem.MolFromSmiles('*c1c(C)cccc1(O)')
chain = Chem.MolFromSmiles('NC')
products = Chem.ReplaceSubstructs(core,Chem.MolFromSmarts('[#0]'),chain) # tuple
print(Chem.MolToSmiles(products[0]))
CNc1c(C)cccc1O
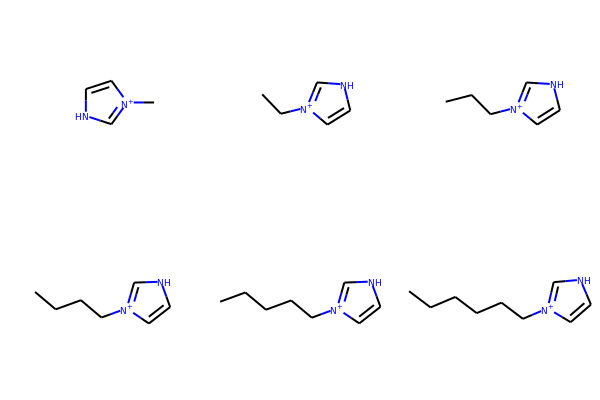
# Here is an example in a loop for an imidazolium core with alkyl chains
core = Chem.MolFromSmiles('*[n+]1cc[nH]c1')
chains = ['C','CC','CCC','CCCC','CCCCC','CCCCCC']
chainMols = [Chem.MolFromSmiles(chain) for chain in chains]
product_smi = []
for chainMol in chainMols:
product_mol = Chem.ReplaceSubstructs(core,Chem.MolFromSmarts('[#0]'),chainMol)
product_smi.append(Chem.MolToSmiles(product_mol[0]))
print(product_smi)
['C[n+]1cc[nH]c1', 'CC[n+]1cc[nH]c1', 'CCC[n+]1cc[nH]c1', 'CCCC[n+]1cc[nH]c1', 'CCCCC[n+]1cc[nH]c1', 'CCCCCC[n+]1cc[nH]c1']
# View the enumerated molecules:
Draw.MolsToGridImage([Chem.MolFromSmiles(smi) for smi in product_smi])
Neutralizing Molecules¶
This neutralize_atoms() algorithm is adapted from Noel O’Boyle’s nocharge code. It is a
neutralization by atom approach and neutralizes atoms with a +1 or -1 charge by removing or
adding hydrogen where possible. The SMARTS pattern checks for a hydrogen in +1 charged atoms and
checks for no neighbors with a negative charge (for +1 atoms) and no neighbors with a positive charge
(for -1 atoms), this is to avoid altering molecules with charge separation (e.g., nitro groups).
The neutralize_atoms() function differs from the rdMolStandardize.Uncharger behavior.
See the MolVS documentation for Uncharger:
https://molvs.readthedocs.io/en/latest/api.html#molvs-charge
“This class uncharges molecules by adding and/or removing hydrogens. In cases where there is a positive charge that is not neutralizable, any corresponding negative charge is also preserved.”
As an example, rdMolStandardize.Uncharger will not change charges on C[N+](C)(C)CCC([O-])=O,
as there is a positive charge that is not neutralizable. In contrast, the neutralize_atoms() function
will attempt to neutralize any atoms it can (in this case to C[N+](C)(C)CCC(=O)O).
That is, neutralize_atoms() ignores the overall charge on the molecule, and attempts to neutralize charges
even if the neutralization introduces an overall formal charge on the molecule. See below for a comparison.
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
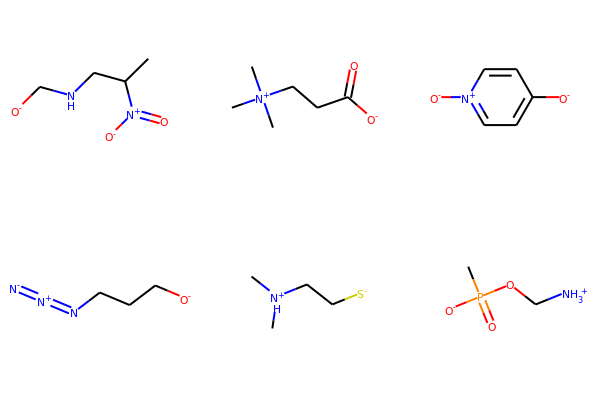
# list of SMILES
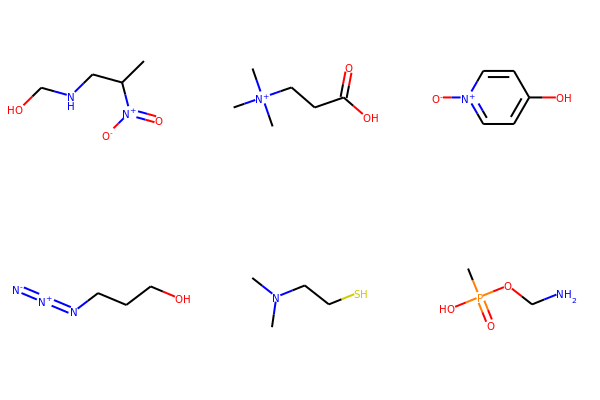
smiList = ['CC(CNC[O-])[N+]([O-])=O',
'C[N+](C)(C)CCC([O-])=O',
'[O-]C1=CC=[N+]([O-])C=C1',
'[O-]CCCN=[N+]=[N-]',
'C[NH+](C)CC[S-]',
'CP([O-])(=O)OC[NH3+]']
# Create RDKit molecular objects
mols = [Chem.MolFromSmiles(m) for m in smiList]
# display
Draw.MolsToGridImage(mols,molsPerRow=3,subImgSize=(200,200))
def neutralize_atoms(mol):
pattern = Chem.MolFromSmarts("[+1!h0!$([*]~[-1,-2,-3,-4]),-1!$([*]~[+1,+2,+3,+4])]")
at_matches = mol.GetSubstructMatches(pattern)
at_matches_list = [y[0] for y in at_matches]
if len(at_matches_list) > 0:
for at_idx in at_matches_list:
atom = mol.GetAtomWithIdx(at_idx)
chg = atom.GetFormalCharge()
hcount = atom.GetTotalNumHs()
atom.SetFormalCharge(0)
atom.SetNumExplicitHs(hcount - chg)
atom.UpdatePropertyCache()
return mol
# Neutralize molecules by atom
for mol in mols:
neutralize_atoms(mol)
print(Chem.MolToSmiles(mol))
CC(CNCO)[N+](=O)[O-]
C[N+](C)(C)CCC(=O)O
[O-][n+]1ccc(O)cc1
[N-]=[N+]=NCCCO
CN(C)CCS
CP(=O)(O)OCN
Draw.MolsToGridImage(mols,molsPerRow=3, subImgSize=(200,200))
Compare to rdMolStandardize.Uncharger results:
from rdkit.Chem.MolStandardize import rdMolStandardize
un = rdMolStandardize.Uncharger()
mols2 = [Chem.MolFromSmiles(m) for m in smiList]
for mol2 in mols2:
un.uncharge(mol2)
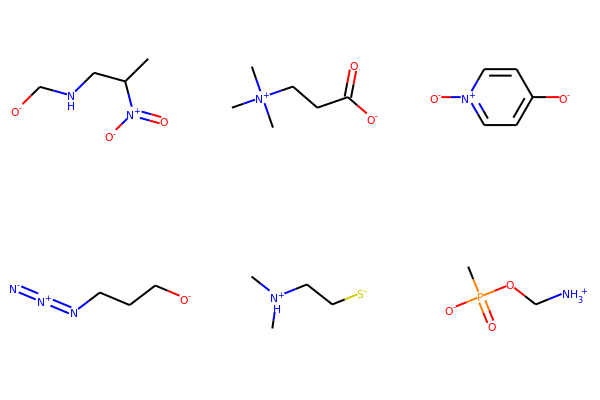
print(Chem.MolToSmiles(mol2))
CC(CNC[O-])[N+](=O)[O-]
C[N+](C)(C)CCC(=O)[O-]
[O-]c1cc[n+]([O-])cc1
[N-]=[N+]=NCCC[O-]
C[NH+](C)CC[S-]
CP(=O)([O-])OC[NH3+]
Draw.MolsToGridImage(mols2,molsPerRow=3,subImgSize=(200,200))
Substructure Matching¶
Functional Group with SMARTS queries¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
sucrose = "C([C@@H]1[C@H]([C@@H]([C@H]([C@H](O1)O[C@]2([C@H]([C@@H]([C@H](O2)CO)O)O)CO)O)O)O)O"
sucrose_mol = Chem.MolFromSmiles(sucrose)
primary_alcohol = Chem.MolFromSmarts("[CH2][OH1]")
print(sucrose_mol.GetSubstructMatches(primary_alcohol))
((0, 22), (13, 14), (17, 18))
secondary_alcohol = Chem.MolFromSmarts("[CH1][OH1]")
print(sucrose_mol.GetSubstructMatches(secondary_alcohol))
((2, 21), (3, 20), (4, 19), (9, 16), (10, 15))
Macrocycles with SMARTS queries¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
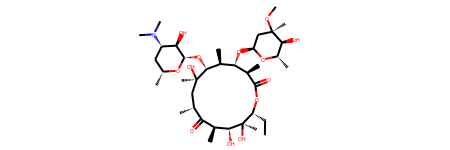
erythromycin = Chem.MolFromSmiles("CC[C@@H]1[C@@]([C@@H]([C@H](C(=O)[C@@H](C[C@@]([C@@H]([C@H]([C@@H]([C@H](C(=O)O1)C)O[C@H]2C[C@@]([C@H]([C@@H](O2)C)O)(C)OC)C)O[C@H]3[C@@H]([C@H](C[C@H](O3)C)N(C)C)O)(C)O)C)C)O)(C)O")
erythromycin
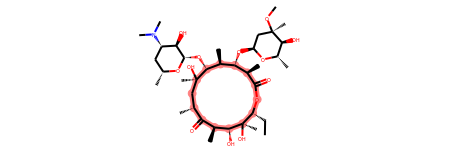
# Define SMARTS pattern with ring size > 12
# This is an RDKit SMARTS extension
macro = Chem.MolFromSmarts("[r{12-}]")
print(erythromycin.GetSubstructMatches(macro))
((2,), (3,), (4,), (5,), (6,), (8,), (9,), (10,), (11,), (12,), (13,), (14,), (15,), (17,))
erythromycin
Returning Substructure Matches as SMILES¶
from rdkit import Chem
pat = Chem.MolFromSmarts("[NX1]#[CX2]") #matches nitrile
mol = Chem.MolFromSmiles("CCCC#N") # Butyronitrile
atom_indices = mol.GetSubstructMatch(pat)
print(atom_indices)
(4, 3)
print(Chem.MolFragmentToSmiles(mol, atom_indices)) # returns the nitrile
C#N
# Note however that if only the atom indices are given then Chem.MolFragmentToSmiles() will include all bonds
# which connect those atoms, even if the original SMARTS does not match those bonds. For example:
pat = Chem.MolFromSmarts("*~*~*~*") # match 4 linear atoms
mol = Chem.MolFromSmiles("C1CCC1") # ring of size 4
atom_indices = mol.GetSubstructMatch(pat)
print(atom_indices)
(0, 1, 2, 3)
print(Chem.MolFragmentToSmiles(mol, atom_indices)) # returns the ring
C1CCC1
# If this is important, then you need to pass the correct bond indices to MolFragmentToSmiles().
# This can be done by using the bonds in the query graph to get the bond indices in the molecule graph.
def get_match_bond_indices(query, mol, match_atom_indices):
bond_indices = []
for query_bond in query.GetBonds():
atom_index1 = match_atom_indices[query_bond.GetBeginAtomIdx()]
atom_index2 = match_atom_indices[query_bond.GetEndAtomIdx()]
bond_indices.append(mol.GetBondBetweenAtoms(
atom_index1, atom_index2).GetIdx())
return bond_indices
bond_indices = get_match_bond_indices(pat, mol, atom_indices)
print(bond_indices)
[0, 1, 2]
print(Chem.MolFragmentToSmiles(mol, atom_indices, bond_indices))
CCCC
Within the Same Fragment¶
from rdkit import Chem
p = Chem.MolFromSmarts('O.N')
# define a function where matches are contained in a single fragment
def fragsearch(m,p):
matches = [set(x) for x in m.GetSubstructMatches(p)]
frags = [set(y) for y in Chem.GetMolFrags(m)] # had to add this line for code to work
for frag in frags:
for match in matches:
if match.issubset(frag):
return match
return False
m1 = Chem.MolFromSmiles('OCCCN.CCC')
m1
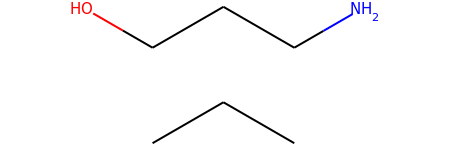
m2 = Chem.MolFromSmiles('OCCC.CCCN')
m2
print(m1.HasSubstructMatch(p))
True
print(m2.HasSubstructMatch(p))
True
print(fragsearch(m1,p))
{0, 4}
print(fragsearch(m2,p))
False
Descriptor Calculations¶
Molecule Hash Strings¶
from rdkit import Chem
from rdkit.Chem import rdMolHash
import rdkit
s = Chem.MolFromSmiles('CC(C(C1=CC(=C(C=C1)O)O)O)N(C)C(=O)OCC2=CC=CC=C2')
s
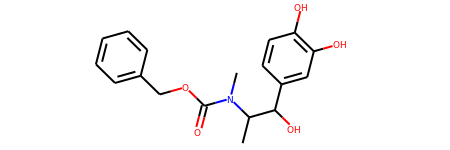
# View all of the MolHash hashing functions types with the names method.
molhashf = rdMolHash.HashFunction.names
print(molhashf)
{'AnonymousGraph': rdkit.Chem.rdMolHash.HashFunction.AnonymousGraph,
'ElementGraph': rdkit.Chem.rdMolHash.HashFunction.ElementGraph,
'CanonicalSmiles': rdkit.Chem.rdMolHash.HashFunction.CanonicalSmiles,
'MurckoScaffold': rdkit.Chem.rdMolHash.HashFunction.MurckoScaffold,
'ExtendedMurcko': rdkit.Chem.rdMolHash.HashFunction.ExtendedMurcko,
'MolFormula': rdkit.Chem.rdMolHash.HashFunction.MolFormula,
'AtomBondCounts': rdkit.Chem.rdMolHash.HashFunction.AtomBondCounts,
'DegreeVector': rdkit.Chem.rdMolHash.HashFunction.DegreeVector,
'Mesomer': rdkit.Chem.rdMolHash.HashFunction.Mesomer,
'HetAtomTautomer': rdkit.Chem.rdMolHash.HashFunction.HetAtomTautomer,
'HetAtomProtomer': rdkit.Chem.rdMolHash.HashFunction.HetAtomProtomer,
'RedoxPair': rdkit.Chem.rdMolHash.HashFunction.RedoxPair,
'Regioisomer': rdkit.Chem.rdMolHash.HashFunction.Regioisomer,
'NetCharge': rdkit.Chem.rdMolHash.HashFunction.NetCharge,
'SmallWorldIndexBR': rdkit.Chem.rdMolHash.HashFunction.SmallWorldIndexBR,
'SmallWorldIndexBRL': rdkit.Chem.rdMolHash.HashFunction.SmallWorldIndexBRL,
'ArthorSubstructureOrder': rdkit.Chem.rdMolHash.HashFunction.ArthorSubstructureOrder,
'HetAtomTautomerv2': rdkit.Chem.rdMolHash.HashFunction.HetAtomTautomerv2,
'HetAtomProtomerv2': rdkit.Chem.rdMolHash.HashFunction.HetAtomProtomerv2}
# Generate MolHashes for molecule 's' with all defined hash functions.
for i, j in molhashf.items():
print(i, rdMolHash.MolHash(s, j))
AnonymousGraph **1***(*(*)*(*)*(*)*(*)***2*****2)**1*
ElementGraph CC(C(O)C1CCC(O)C(O)C1)N(C)C(O)OCC1CCCCC1
CanonicalSmiles CC(C(O)c1ccc(O)c(O)c1)N(C)C(=O)OCc1ccccc1
MurckoScaffold c1ccc(CCNCOCc2ccccc2)cc1
ExtendedMurcko *c1ccc(C(*)C(*)N(*)C(=*)OCc2ccccc2)cc1*
MolFormula C18H21NO5
AtomBondCounts 24,25
DegreeVector 0,8,10,6
Mesomer CC(C(O)[C]1[CH][CH][C](O)[C](O)[CH]1)N(C)[C]([O])OC[C]1[CH][CH][CH][CH][CH]1_0
HetAtomTautomer CC(C([O])[C]1[CH][CH][C]([O])[C]([O])[CH]1)N(C)[C]([O])OC[C]1[CH][CH][CH][CH][CH]1_3_0
HetAtomProtomer CC(C([O])[C]1[CH][CH][C]([O])[C]([O])[CH]1)N(C)[C]([O])OC[C]1[CH][CH][CH][CH][CH]1_3
RedoxPair CC(C(O)[C]1[CH][CH][C](O)[C](O)[CH]1)N(C)[C]([O])OC[C]1[CH][CH][CH][CH][CH]1
Regioisomer *C.*CCC.*O.*O.*O.*OC(=O)N(*)*.C.c1ccccc1.c1ccccc1
NetCharge 0
SmallWorldIndexBR B25R2
SmallWorldIndexBRL B25R2L10
ArthorSubstructureOrder 00180019010012000600009b000000
HetAtomTautomerv2 [CH3]-[CH](-[CH](-[OH])-[C]1:[C]:[C]:[C](:[O]):[C](:[O]):[C]:1)-[N](-[CH3])-[C](=[O])-[O]-[CH2]-[c]1:[cH]:[cH]:[cH]:[cH]:[cH]:1_5_0
HetAtomProtomerv2 [CH3]-[CH](-[CH](-[OH])-[C]1:[C]:[C]:[C](:[O]):[C](:[O]):[C]:1)-[N](-[CH3])-[C](=[O])-[O]-[CH2]-[c]1:[cH]:[cH]:[cH]:[cH]:[cH]:1_5
# Murcko Scaffold Hashes (from slide 16 in [ref2])
# Create a list of SMILES
mList = ['CCC1CC(CCC1=O)C(=O)C1=CC=CC(C)=C1','CCC1CC(CCC1=O)C(=O)C1=CC=CC=C1',\
'CC(=C)C(C1=CC=CC=C1)S(=O)CC(N)=O','CC1=CC(=CC=C1)C(C1CCC(N)CC1)C(F)(F)F',\
'CNC1CCC(C2=CC(Cl)=C(Cl)C=C2)C2=CC=CC=C12','CCCOC(C1CCCCC1)C1=CC=C(Cl)C=C1']
# Loop through the SMILES mList and create RDKit molecular objects
mMols = [Chem.MolFromSmiles(m) for m in mList]
# Calculate Murcko Scaffold Hashes
murckoHashList = [rdMolHash.MolHash(mMol, rdkit.Chem.rdMolHash.HashFunction.MurckoScaffold) for mMol in mMols]
print(murckoHashList)
['c1ccc(CC2CCCCC2)cc1',
'c1ccc(CC2CCCCC2)cc1',
'c1ccccc1',
'c1ccc(CC2CCCCC2)cc1',
'c1ccc(C2CCCc3ccccc32)cc1',
'c1ccc(CC2CCCCC2)cc1']
# Get the most frequent Murcko Scaffold Hash
def mostFreq(list):
return max(set(list), key=list.count)
mostFreq_murckoHash = mostFreq(murckoHashList)
print(mostFreq_murckoHash)
c1ccc(CC2CCCCC2)cc1
mostFreq_murckoHash_mol = Chem.MolFromSmiles('c1ccc(CC2CCCCC2)cc1')
mostFreq_murckoHash_mol
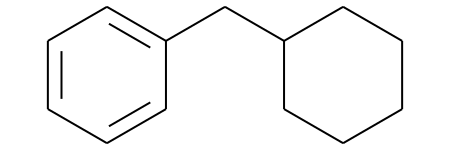
# Display molecules with MurkoHash as legends and highlight the mostFreq_murckoHash
highlight_mostFreq_murckoHash = [mMol.GetSubstructMatch(mostFreq_murckoHash_mol) for mMol in mMols]
Draw.MolsToGridImage(mMols,legends=[murckoHash for murckoHash in murckoHashList],
highlightAtomLists = highlight_mostFreq_murckoHash,
subImgSize=(250,250), useSVG=False)
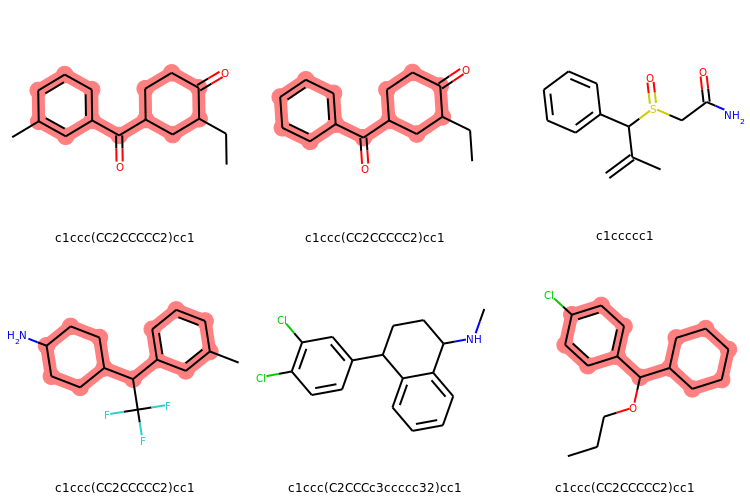
# Regioisomer Hashes (from slide 17 in [ref2])
# Find Regioisomer matches for this molecule
r0 = Chem.MolFromSmiles('CC1=CC2=C(C=C1)C(=CN2CCN1CCOCC1)C(=O)C1=CC=CC2=C1C=CC=C2')
r0
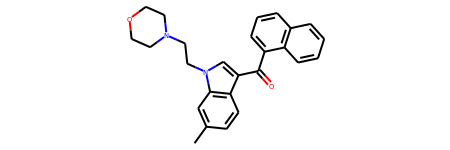
# Calculate the regioisomer hash for r0
r0_regioHash = rdMolHash.MolHash(r0,rdkit.Chem.rdMolHash.HashFunction.Regioisomer)
print(r0_regioHash)
*C.*C(*)=O.*CC*.C1COCCN1.c1ccc2[nH]ccc2c1.c1ccc2ccccc2c1
r0_regioHash_mol = Chem.MolFromSmiles('*C.*C(*)=O.*CC*.C1COCCN1.c1ccc2[nH]ccc2c1.c1ccc2ccccc2c1')
r0_regioHash_mol
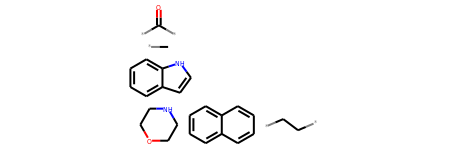
# Create a list of SMILES
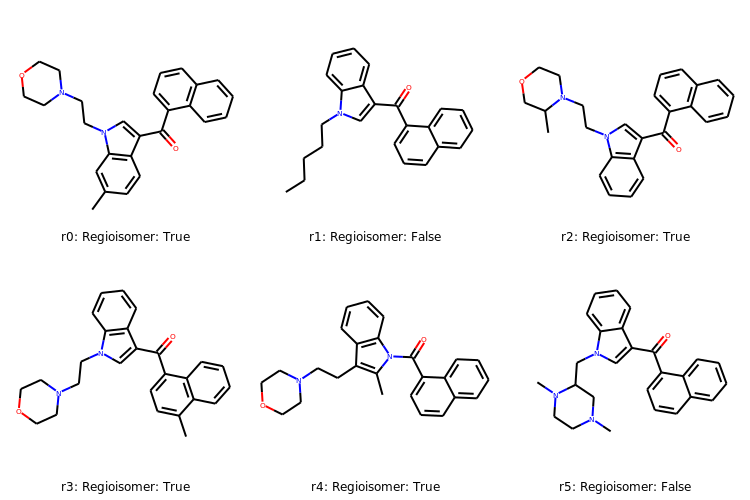
rList = ['CC1=CC2=C(C=C1)C(=CN2CCN1CCOCC1)C(=O)C1=CC=CC2=C1C=CC=C2',\
'CCCCCN1C=C(C2=CC=CC=C21)C(=O)C3=CC=CC4=CC=CC=C43',\
'CC1COCCN1CCN1C=C(C(=O)C2=CC=CC3=C2C=CC=C3)C2=C1C=CC=C2',\
'CC1=CC=C(C(=O)C2=CN(CCN3CCOCC3)C3=C2C=CC=C3)C2=C1C=CC=C2',\
'CC1=C(CCN2CCOCC2)C2=C(C=CC=C2)N1C(=O)C1=CC=CC2=CC=CC=C12',\
'CN1CCN(C(C1)CN2C=C(C3=CC=CC=C32)C(=O)C4=CC=CC5=CC=CC=C54)C']
# Loop through the SMILES rList and create RDKit molecular objects
rMols = [Chem.MolFromSmiles(r) for r in rList]
# Calculate Regioisomer Hashes
regioHashList = [rdMolHash.MolHash(rMol, rdkit.Chem.rdMolHash.HashFunction.Regioisomer) for rMol in rMols]
print(regioHashList)
['*C.*C(*)=O.*CC*.C1COCCN1.c1ccc2[nH]ccc2c1.c1ccc2ccccc2c1',
'*C(*)=O.*CCCCC.c1ccc2[nH]ccc2c1.c1ccc2ccccc2c1',
'*C.*C(*)=O.*CC*.C1COCCN1.c1ccc2[nH]ccc2c1.c1ccc2ccccc2c1',
'*C.*C(*)=O.*CC*.C1COCCN1.c1ccc2[nH]ccc2c1.c1ccc2ccccc2c1',
'*C.*C(*)=O.*CC*.C1COCCN1.c1ccc2[nH]ccc2c1.c1ccc2ccccc2c1',
'*C.*C.*C(*)=O.*C*.C1CNCCN1.c1ccc2[nH]ccc2c1.c1ccc2ccccc2c1']
rmatches =[]
for regioHash in regioHashList:
if regioHash == r0_regioHash:
print('Regioisomer: True')
rmatches.append('Regioisomer: True')
else:
print('Regioisomer: False')
rmatches.append('Regioisomer: False')
Regioisomer: True
Regioisomer: False
Regioisomer: True
Regioisomer: True
Regioisomer: True
Regioisomer: False
# Create some labels
index = ['r0: ','r1: ','r2: ','r3: ','r4: ','r5: ']
labelList = [rmatches + index for rmatches,index in zip(index,rmatches)]
# Display molecules with labels
Draw.MolsToGridImage(rMols,legends=[label for label in labelList],
subImgSize=(250,250), useSVG=False)
# note, that r0 is the initial molecule we were interested in.
Contiguous Rotatable Bonds¶
from rdkit import Chem
from rdkit.Chem.Lipinski import RotatableBondSmarts
mol = Chem.MolFromSmiles('CCC(CC(C)CC1CCC1)C(CC(=O)O)N')
mol
def find_bond_groups(mol):
"""Find groups of contiguous rotatable bonds and return them sorted by decreasing size"""
rot_atom_pairs = mol.GetSubstructMatches(RotatableBondSmarts)
rot_bond_set = set([mol.GetBondBetweenAtoms(*ap).GetIdx() for ap in rot_atom_pairs])
rot_bond_groups = []
while (rot_bond_set):
i = rot_bond_set.pop()
connected_bond_set = set([i])
stack = [i]
while (stack):
i = stack.pop()
b = mol.GetBondWithIdx(i)
bonds = []
for a in (b.GetBeginAtom(), b.GetEndAtom()):
bonds.extend([b.GetIdx() for b in a.GetBonds() if (
(b.GetIdx() in rot_bond_set) and (not (b.GetIdx() in connected_bond_set)))])
connected_bond_set.update(bonds)
stack.extend(bonds)
rot_bond_set.difference_update(connected_bond_set)
rot_bond_groups.append(tuple(connected_bond_set))
return tuple(sorted(rot_bond_groups, reverse = True, key = lambda x: len(x)))
# Find groups of contiguous rotatable bonds in mol
bond_groups = find_bond_groups(mol)
# As bond groups are sorted by decreasing size, the size of the first group (if any)
# is the largest number of contiguous rotatable bonds in mol
largest_n_cont_rot_bonds = len(bond_groups[0]) if bond_groups else 0
print(largest_n_cont_rot_bonds)
8
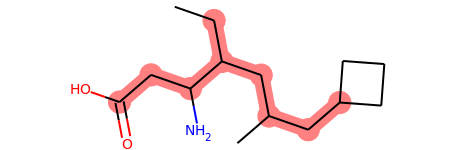
print(bond_groups)
((1, 2, 3, 5, 6, 10, 11, 12),)
mol
Writing Molecules¶
Kekule SMILES¶
from rdkit import Chem
smi = "CN1C(NC2=NC=CC=C2)=CC=C1"
mol = Chem.MolFromSmiles(smi)
print(Chem.MolToSmiles(mol))
Cn1cccc1Nc1ccccn1
Chem.Kekulize(mol)
print(Chem.MolToSmiles(mol, kekuleSmiles=True))
CN1C=CC=C1NC1=CC=CC=N1
Isomeric SMILES without isotopes¶
from rdkit import Chem
def MolWithoutIsotopesToSmiles(mol):
atom_data = [(atom, atom.GetIsotope()) for atom in mol.GetAtoms()]
for atom, isotope in atom_data:
# restore original isotope values
if isotope:
atom.SetIsotope(0)
smiles = Chem.MolToSmiles(mol)
for atom, isotope in atom_data:
if isotope:
atom.SetIsotope(isotope)
return smiles
mol = Chem.MolFromSmiles("[19F][13C@H]([16OH])[35Cl]")
print(MolWithoutIsotopesToSmiles(mol))
O[C@@H](F)Cl
N.B. There are two limitations noted with this Isomeric SMILES without isotopes method including with isotopic hydrogens, and a requirement to recalculate stereochemistry. See the source discussion linked above for further explanation and examples.
Reactions¶
Reversing Reactions¶
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
# Pictet-Spengler rxn
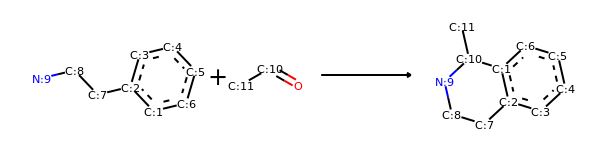
rxn = AllChem.ReactionFromSmarts('[cH1:1]1:[c:2](-[CH2:7]-[CH2:8]-[NH2:9]):[c:3]:[c:4]:[c:5]:[c:6]:1.[#6:11]-[CH1;R0:10]=[OD1]>>[c:1]12:[c:2](-[CH2:7]-[CH2:8]-[NH1:9]-[C:10]-2(-[#6:11])):[c:3]:[c:4]:[c:5]:[c:6]:1')
rxn
rxn2 = AllChem.ChemicalReaction()
for i in range(rxn.GetNumReactantTemplates()):
rxn2.AddProductTemplate(rxn.GetReactantTemplate(i))
for i in range(rxn.GetNumProductTemplates()):
rxn2.AddReactantTemplate(rxn.GetProductTemplate(i))
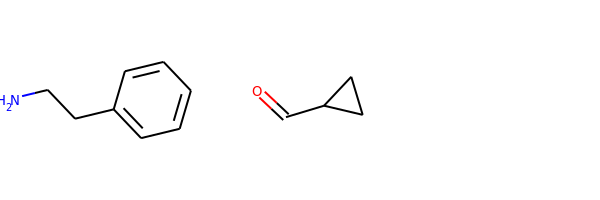
rxn2.Initialize()
reacts = [Chem.MolFromSmiles(x) for x in ('NCCc1ccccc1','C1CC1C(=O)')]
ps = rxn.RunReactants(reacts)
ps0 = ps[0]
for p in ps0:
Chem.SanitizeMol(p)
Draw.MolsToGridImage(ps0)

reacts = ps0
rps = rxn2.RunReactants(reacts)
rps0 = rps[0]
for rp in rps0:
Chem.SanitizeMol(rp)
Draw.MolsToGridImage(rps0)
N.B. This approach isn’t perfect and won’t work for every reaction. Reactions that include extensive query information in the original reactants are very likely to be problematic.
Reaction Fingerprints and Similarity¶
from rdkit import Chem
from rdkit.Chem import rdChemReactions
from rdkit.Chem import DataStructs
# construct the chemical reactions
rxn1 = rdChemReactions.ReactionFromSmarts('CCCO>>CCC=O')
rxn2 = rdChemReactions.ReactionFromSmarts('CC(O)C>>CC(=O)C')
rxn3 = rdChemReactions.ReactionFromSmarts('NCCO>>NCC=O')
# construct difference fingerprint (subtracts reactant fingerprint from product)
fp1 = rdChemReactions.CreateDifferenceFingerprintForReaction(rxn1)
fp2 = rdChemReactions.CreateDifferenceFingerprintForReaction(rxn2)
fp3 = rdChemReactions.CreateDifferenceFingerprintForReaction(rxn3)
print(DataStructs.TanimotoSimilarity(fp1,fp2))
0.0
# The similarity between fp1 and fp2 is zero because as far as the reaction
# fingerprint is concerned, the parts which change within the reactions have
# nothing in common with each other.
# In contrast, fp1 and fp3 have some common parts
print(DataStructs.TanimotoSimilarity(fp1,fp3))
0.42857142857142855
Error Messages¶
Explicit Valence Error - Partial Sanitization¶
from rdkit import Chem
from rdkit.Chem import rdqueries
The default RDKit behavior is to reject hypervalent P, so you need to set sanitize=False:
m = Chem.MolFromSmiles('F[P-](F)(F)(F)(F)F.CN(C)C(F)=[N+](C)C',sanitize=False)
m
The arrangement of the six F around the P is not the octahedral arrangement we would expect because the RDKit has not assigned a hybridization to the P (or any other atoms):
# Build a query for the P
q = rdqueries.AtomNumEqualsQueryAtom(15)
# Select the first and only P
phosphorus = m.GetAtomsMatchingQuery(q)[0]
print(phosphorus.GetHybridization())
UNSPECIFIED
Next, you probably want to at least do a partial sanitization so that the molecule is actually useful. In this case, setting the hybridization is key:
# Regenerate computed properties like implicit valence and ring information
m.UpdatePropertyCache(strict=False)
# Apply several sanitization rules
Chem.SanitizeMol(m,Chem.SanitizeFlags.SANITIZE_FINDRADICALS|Chem.SanitizeFlags.SANITIZE_KEKULIZE|Chem.SanitizeFlags.SANITIZE_SETAROMATICITY|Chem.SanitizeFlags.SANITIZE_SETCONJUGATION|Chem.SanitizeFlags.SANITIZE_SETHYBRIDIZATION|Chem.SanitizeFlags.SANITIZE_SYMMRINGS,catchErrors=True)
m
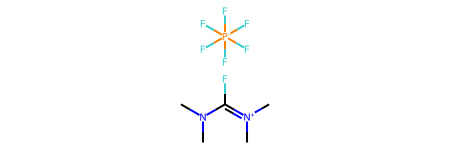
Now the expected octahedral arrangement of the six F around the P exists because the hybridization of P has been assigned as SP3D2:
print(phosphorus.GetHybridization())
SP3D2
Detect Chemistry Problems¶
from rdkit import Chem
m = Chem.MolFromSmiles('CN(C)(C)C', sanitize=False)
problems = Chem.DetectChemistryProblems(m)
print(len(problems))
1
print(problems[0].GetType())
print(problems[0].GetAtomIdx())
print(problems[0].Message())
AtomValenceException
1
Explicit valence for atom # 1 N, 4, is greater than permitted
m2 = Chem.MolFromSmiles('c1cncc1',sanitize=False)
problems = Chem.DetectChemistryProblems(m2)
print(len(problems))
1
print(problems[0].GetType())
print(problems[0].GetAtomIndices())
print(problems[0].Message())
KekulizeException
(0, 1, 2, 3, 4)
Can't kekulize mol. Unkekulized atoms: 0 1 2 3 4
Miscellaneous Topics¶
Explicit Valence and Number of Hydrogens¶
Most of the time (exception is explained below), explicit refers to atoms that are in the graph and implicit refers to atoms that are not in the graph (i.e., Hydrogens). So given that the ring is aromatic (e.g.,in pyrrole), the explicit valence of each of the atoms (ignoring the Hs that are not present in the graph) in pyrrole is 3. If you want the Hydrogen count, use GetTotalNumHs(); the total number of Hs for each atom is one:
from rdkit import Chem
pyrrole = Chem.MolFromSmiles('C1=CNC=C1')
for atom in pyrrole.GetAtoms():
print(atom.GetSymbol(), atom.GetExplicitValence(), atom.GetTotalNumHs())
C 3 1
C 3 1
N 3 1
C 3 1
C 3 1
In RDKit, there is overlapping nomenclature around the use of the words “explicit” and “implicit” when it comes to Hydrogens. When you specify the Hydrogens for an atom inside of square brackets in the SMILES, it becomes an “explicit” hydrogen as far as atom.GetNumExplicitHs() is concerned. Here is an example:
pyrrole = Chem.MolFromSmiles('C1=CNC=C1')
mol1 = Chem.MolFromSmiles('C1=CNCC1')
mol2 = Chem.MolFromSmiles('C1=C[NH]CC1')
for atom in pyrrole.GetAtoms():
print(atom.GetSymbol(), atom.GetExplicitValence(), atom.GetNumImplicitHs(), atom.GetNumExplicitHs(), atom.GetTotalNumHs())
C 3 1 0 1
C 3 1 0 1
N 3 0 1 1
C 3 1 0 1
C 3 1 0 1
for atom in mol1.GetAtoms():
print(atom.GetSymbol(), atom.GetExplicitValence(), atom.GetNumImplicitHs(), atom.GetNumExplicitHs(), atom.GetTotalNumHs())
C 3 1 0 1
C 3 1 0 1
N 2 1 0 1
C 2 2 0 2
C 2 2 0 2
for atom in mol2.GetAtoms():
print(atom.GetSymbol(), atom.GetExplicitValence(), atom.GetNumImplicitHs(), atom.GetNumExplicitHs(), atom.GetTotalNumHs())
C 3 1 0 1
C 3 1 0 1
N 3 0 1 1
C 2 2 0 2
C 2 2 0 2
Wiener Index¶
from rdkit import Chem
def wiener_index(m):
res = 0
amat = Chem.GetDistanceMatrix(m)
num_atoms = m.GetNumAtoms()
for i in range(num_atoms):
for j in range(i+1,num_atoms):
res += amat[i][j]
return res
butane = Chem.MolFromSmiles('CCCC')
print(wiener_index(butane))
10.0
isobutane = Chem.MolFromSmiles('CC(C)C')
print(wiener_index(isobutane))
9.0
Organometallics with Dative Bonds¶
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
def is_transition_metal(at):
n = at.GetAtomicNum()
return (n>=22 and n<=29) or (n>=40 and n<=47) or (n>=72 and n<=79)
def set_dative_bonds(mol, fromAtoms=(7,8)):
""" convert some bonds to dative
Replaces some single bonds between metals and atoms with atomic numbers in fomAtoms
with dative bonds. The replacement is only done if the atom has "too many" bonds.
Returns the modified molecule.
"""
pt = Chem.GetPeriodicTable()
rwmol = Chem.RWMol(mol)
rwmol.UpdatePropertyCache(strict=False)
metals = [at for at in rwmol.GetAtoms() if is_transition_metal(at)]
for metal in metals:
for nbr in metal.GetNeighbors():
if nbr.GetAtomicNum() in fromAtoms and \
nbr.GetExplicitValence()>pt.GetDefaultValence(nbr.GetAtomicNum()) and \
rwmol.GetBondBetweenAtoms(nbr.GetIdx(),metal.GetIdx()).GetBondType() == Chem.BondType.SINGLE:
rwmol.RemoveBond(nbr.GetIdx(),metal.GetIdx())
rwmol.AddBond(nbr.GetIdx(),metal.GetIdx(),Chem.BondType.DATIVE)
return rwmol
m = Chem.MolFromSmiles('CN(C)(C)[Pt]', sanitize=False)
m2 = set_dative_bonds(m)
m2
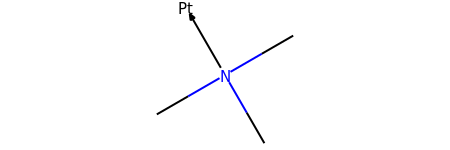
# we can check the bond between nitrogen and platinum
print(m2.GetBondBetweenAtoms(1,4).GetBondType())
DATIVE
# It also shows up in the output SMILES
# This is an RDKit extension to SMILES
print(Chem.MolToSmiles(m2))
C[N](C)(C)->[Pt]
Enumerate SMILES¶
from rdkit import Chem
# create a mol object
mol = Chem.MolFromSmiles('CC(N)C1CC1')
# Generate 100 random SMILES
smis = []
for i in range(100):
smis.append(Chem.MolToSmiles(mol,doRandom=True,canonical=False))
# remove duplicates
smis_set = list(set(smis))
print(smis_set) # output order will be random; doctest skipped
['NC(C)C1CC1',
'C1(C(N)C)CC1',
'C(N)(C)C1CC1',
'CC(C1CC1)N',
'C1C(C(N)C)C1',
'C1C(C1)C(N)C',
'C(C1CC1)(C)N',
'C1(CC1)C(C)N',
'C1C(C(C)N)C1',
'C1CC1C(C)N',
'C(C1CC1)(N)C',
'C1(C(C)N)CC1',
'C1C(C1)C(C)N',
'C(C)(C1CC1)N',
'C1CC1C(N)C',
'C1(CC1)C(N)C',
'C(N)(C1CC1)C',
'NC(C1CC1)C',
'CC(N)C1CC1',
'C(C)(N)C1CC1']
# If you need the multiple random SMILES strings to be reproducible,
# the 2020.09 release has an option for this:
m = Chem.MolFromSmiles('Oc1ncc(OC(CC)C)cc1')
print(Chem.MolToRandomSmilesVect(m,5)) # output order random; doctest skipped
['c1c(cnc(O)c1)OC(CC)C', 'c1c(cnc(c1)O)OC(CC)C', 'c1cc(O)ncc1OC(CC)C', 'O(C(CC)C)c1ccc(nc1)O', 'O(C(C)CC)c1cnc(cc1)O']
# by default the results are not reproducible:
print(Chem.MolToRandomSmilesVect(m,5)) # output order random; doctest skipped
['c1nc(O)ccc1OC(CC)C', 'n1cc(OC(CC)C)ccc1O', 'c1c(OC(C)CC)ccc(O)n1', 'CCC(Oc1ccc(nc1)O)C', 'O(c1cnc(cc1)O)C(C)CC']
# But we can provide a random number seed:
m = Chem.MolFromSmiles('Oc1ncc(OC(CC)C)cc1')
s1 = Chem.MolToRandomSmilesVect(m,5,randomSeed=0xf00d)
print(s1)
['Oc1ccc(OC(CC)C)cn1', 'CC(CC)Oc1cnc(O)cc1', 'c1(O)ncc(cc1)OC(C)CC', 'c1cc(cnc1O)OC(CC)C', 'c1c(OC(CC)C)cnc(c1)O']
s2 = Chem.MolToRandomSmilesVect(m,5,randomSeed=0xf00d)
print(s2 == s1)
True
Reorder Atoms¶
from rdkit import Chem
from rdkit.Chem.Draw import MolsToGridImage
m = Chem.MolFromSmiles("c1([C@H](C)CC)cccc2ccccc12")
m1 = Chem.MolFromSmiles("c12ccccc1c(ccc2)[C@H](C)CC")
print(Chem.MolToSmiles(m) == Chem.MolToSmiles(m1))
True
# check if current canonical atom ordering matches
m_neworder = tuple(zip(*sorted([(j, i) for i, j in enumerate(Chem.CanonicalRankAtoms(m))])))[1]
m1_neworder = tuple(zip(*sorted([(j, i) for i, j in enumerate(Chem.CanonicalRankAtoms(m1))])))[1]
print(m_neworder == m1_neworder)
False
# add atom numbers in images
def addAtomIndices(mol):
for i, a in enumerate(mol.GetAtoms()):
a.SetAtomMapNum(i)
addAtomIndices(m)
addAtomIndices(m1)
MolsToGridImage((m, m1))
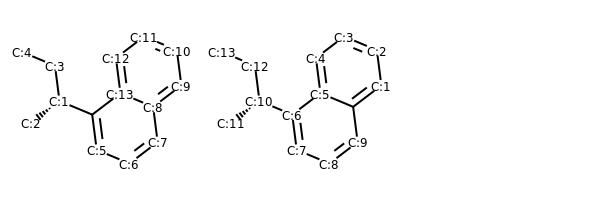
# renumber atoms with same canonical ordering
m_renum = Chem.RenumberAtoms(m, m_neworder)
m1_renum = Chem.RenumberAtoms(m1, m1_neworder)
addAtomIndices(m_renum)
addAtomIndices(m1_renum)
MolsToGridImage((m_renum, m1_renum))
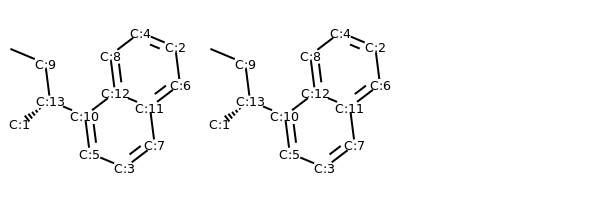
Conformer Generation with ETKDG¶
from rdkit import Chem
from rdkit.Chem import AllChem
To yield more chemically meaningful conformers, Riniker and Landrum implemented the experimental torsion knowledge distance geometry (ETKDG) method [3] which uses torsion angle preferences from the Cambridge Structural Database (CSD) to correct the conformers after distance geometry has been used to generate them. The configs of various conformer generation options are stored in a EmbedParameter object. To explicitly call the ETKDG EmbedParameter object:
params = AllChem.ETKDG()
At the moment this is the default conformer generation routine in RDKit. A newer set of torsion angle potentials were published in 2016 [4], to use these instead:
params = AllChem.ETKDGv2()
In 2020, we devised some improvements to the ETKDG method for sampling small rings and macrocycles [5].
# this includes addtional small ring torsion potentials
params = AllChem.srETKDGv3()
# this includes additional macrocycle ring torsion potentials and macrocycle-specific handles
params = AllChem.ETKDGv3()
# to use the two in conjunction, do:
params = AllChem.ETKDGv3()
params.useSmallRingTorsions = True
# a macrocycle attached to a small ring
mol = Chem.MolFromSmiles("C(OC(CCCCCCC(OCCSC(CCCCCC1)=O)=O)OCCSC1=O)N1CCOCC1")
mol = Chem.AddHs(mol)
AllChem.EmbedMultipleConfs(mol, numConfs = 3 , params = params)
One additional tool we used in the paper is changing the bounds matrix of a molecule during distance geometry. The following code modifies the default molecular bounds matrix, with the idea of confining the conformational space of the molecule:
from rdkit.Chem import rdDistGeom
import rdkit.DistanceGeometry as DG
mol = Chem.MolFromSmiles("C1CCC1C")
mol = Chem.AddHs(mol)
bm = rdDistGeom.GetMoleculeBoundsMatrix(mol)
bm[0,3] = 1.21
bm[3,0] = 1.20
bm[2,3] = 1.21
bm[3,2] = 1.20
bm[4,3] = 1.21
bm[3,4] = 1.20
DG.DoTriangleSmoothing(bm)
params.SetBoundsMat(bm)
Another tool we introduced is setting custom pairwise Coulombic interactions (CPCIs), which mimics additional electrostatic interactions between atom pairs to refine the embedded conformers. The setter takes in a dictionary of integer tuples as keys and reals as values. The following one-liner sets a repulsive (+ve) interaction of strength 0.9 e^2 between the atom indexed 0 and indexed 3, with the idea of keeping these two atoms further apart.
params.SetCPCI({ (0,3) : 0.9 } )
To use the EmbedParameter for conformer generation:
params.useRandomCoords = True
# Note this is only an illustrative example, hydrogens are not added before conformer generation to keep the indices apparant
AllChem.EmbedMultipleConfs(mol, numConfs = 3 , params = params)
Both of these setters can be used to help sampling all kinds of molecules as the users see fit. Nevertheless, to facilitate using them in conformer generation of macrocycles, we devised the python package github.com/rinikerlab/cpeptools to provide chemcially intuitive bound matrices and CPCIs for macrocycles. Example usage cases are shown in the README.
References
License¶

This document is copyright (C) 2007-2020 by Greg Landrum and Vincent Scalfani.
This work is licensed under the Creative Commons Attribution-ShareAlike 4.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-sa/4.0/ or send a letter to Creative Commons, 543 Howard Street, 5th Floor, San Francisco, California, 94105, USA.
The intent of this license is similar to that of the RDKit itself. In simple words: “Do whatever you want with it, but please give us some credit.”